Jordan T. Detamore Week 4
Purpose
The purpose of this study was to continue to learn how to use tools from the Biology Workbench and work with HIV sequences. We will use the tools learned in our own projects
Methods & Results
Activity II-Part I
- The preloaded sequence feels were uploaded into the nucleic acid tool set of the Biology Workbench
- A multiple sequence alignment and distance tree was created from 3 clones from 4 subjects-The subjects, clone, and tree is shown below


- Do clones from each subject cluster together?
- Yes each set of three clones is clustered together
- Do some subjects' clones show more diversity than others?
- Yes, specifically subject 15 had diverse clones while the others seemed to have a lack of diversity
- Brief Description
- The clones of subject 13 were most different from the rest of the subjects
- The clones of subject 15 had the most diversity amongst clones
- The clones of subject 14 were most closely related to the other subjects
Activity II-Part II
- Align all the clones from one subject and calculate S from the number of positions where there is at least one nucleotide distance-repeat for other two clones
- Calculate theta from
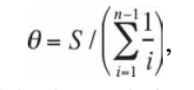
- Theta is an estimate of the average pairwise genetic distance
- Use the Clustdist tool to generate a distance matrix for the alignments
- Select the highest and lowest pairwise scores and convert that percentage different score into the raw number by multiplying by the length of the sequence
- Results from all these tools are listed here:

- Maximum and minimum differences between subjects sequences were then calculated

Data & Files

Conclusion
The main findings from today were the tools learned from completing this task. I learned how to generate S, theta, and minimum and maximum differences. I also gained new tools in analyzing unrolled trees. These tools will be helpful in continuing to pursue the partnered research project.
Defining Research Project
Question
Is there a genetic difference in HIV-1 in patients who contracted the virus sexually or by intravenous injection?
Hypothesis
We hypothesize that there will not be a genetic difference in HIV-1 in patients who contracted the virus sexually or by intravenous injection
Subjects, Visits, Clones
We will use 25 sequences from the moderate progressor category in Markham et al. This will be the best group to compare to outside sequences because they are the most stable group in terms of visits. For the other half of the sequences, we will search more recent studies for sexually contracted HIV-1 sequences. This will be completed during class on 9/26.
Acknowledgements
- I worked with my lab partner Matthew K. Oki in class on this assignment. We shared the research section of the wiki because we are doing the project together
- I also used his tables and figures because we worked with the same subjects and clones in class
- I received instructions and guidance on how to complete this assignment from Dr. Dahlquist's Bioinformatics Lab
- While I worked with the people noted above, this individual journal entry was completed by me and not copied from another source.
Jordan T. Detamore 23:37, 26 September 2016 (EDT):
References
Markham, R.B., Wang, W.C., Weisstein, A.E., Wang, Z., Munoz, A., Templeton, A., Margolick, J., Vlahov, D., Quinn, T., Farzadegan, H., & Yu, X.F. (1998). Patterns of HIV-1 evolution in individuals with differing rates of CD4 T cell decline. Proc Natl Acad Sci U S A. 95, 12568-12573. doi: 10.1073/pnas.95.21.12568
Useful links
LMU Seaver College of Science and Engineering
