BIOL368/F16:Week 15

This journal entry is due on Tuesday, December 13 at midnight PDT (Monday night/Tuesday morning). NOTE that the server records the time as Eastern Daylight Time (EDT). Therefore, midnight will register as 03:00.
Background
References
Background Reading on Microarrays
- Brown, P.O. & Botstein, D. (1999) Exploring the new world of the genome with DNA microarrays Nature Genetics 21: 33-37.
- Campbell, A.M. and Heyer, L.J. (2003), “Chapter 4: Basic Research with DNA Microarrays”, in Discovering Genomics, Proteomics, and Bioinformatics, Cold Spring Harbor Laboratory Press, pp. 107-124. (Available on MyLMUConnect)
- Ernst, J., & Bar-Joseph, Z. (2006). STEM: a tool for the analysis of short time series gene expression data. BMC bioinformatics, 7(1), 1. DOI: 10.1186/1471-2105-7-191
Journal Club Papers
- Hebly, M., de Ridder, D., de Hulster, E. A., de la Torre Cortes, P., Pronk, J. T., & Daran-Lapujade, P. (2014). Physiological and transcriptional responses of anaerobic chemostat cultures of Saccharomyces cerevisiae subjected to diurnal temperature cycles. Applied and environmental microbiology, 80(14), 4433-4449. doi: 10.1128/AEM.00785-14
- Schade, B., Jansen, G., Whiteway, M., Entian, K. D., & Thomas, D. Y. (2004). Cold Adaptation in Budding Yeast. Molecular Biology of the Cell, 15(12), 5492-5502. doi: 10.1091/mbc.E04-03-0167
- Tai, S. L., Daran-Lapujade, P., Walsh, M. C., Pronk, J. T., & Daran, J. M. (2007). Acclimation of Saccharomyces cerevisiae to low temperature: a chemostat-based transcriptome analysis. Molecular Biology of the Cell, 18(12), 5100-5112. doi: 10.1091/mbc.E07-02-0131
Overview of DNA Microarray Analysis
This is a list of steps needed to analyze DNA microarray data.
- Quantitate the fluorescence signal in each spot (GenePix Pro)
- Calculate the ratio of red/green fluorescence (GenePix Pro)
- Log transform the ratios (GenePix Pro)
- Normalize the ratios on each microarray slide (within-chip normalization)
- Normalize the ratios for a set of slides in an experiment (between-chip normalization)
- These first four steps have been done for you.
- Perform statistical analysis on the ratios
- Within-strain ANOVA
- Benjamini & Hochberg and Bonferroni p value corrections
- "Sanity Check"
- Within-strain ANOVA
- Pattern finding algorithms (clustering with stem)
- Gene Ontology term enrichment analysis (on clusters with stem)
Individual Journal Assignment
- Store this journal entry as "username Week 15" (i.e., this is the text to place between the square brackets when you link to this page).
- Create the following set of links. These links should all be in your personal template; then use the template on your journal entry.
- Link to your journal entry from your user page.
- Link back from your journal entry to your user page.
- Link to this assignment from your journal entry.
- Don't forget to add the "BIOL368/F16" category to the end of your wiki page.
Homework Partners
You will work in the same groups as last week.
- Matt A., Matt O., Mia, Colin: Hebly (2014); Δswi4 Dahlquist lab data
- Shivum, Isai, Anu, Will: Tai et al. (2007); Δgln3 Dahlquist lab data
- Avery, Courtney, Jordan, Zach: Schade et al. (2004); wt Dahlquist lab data
Grading
- The Week 15 individual journal (electronic lab notebook) is worth 10 points.
- The Week 15 shared journal is worth 3 points.
- The combined journal club/research group presentation will be worth 50 points.
Electronic Lab Notebook
Complete your electronic notebook that gives the details of what you did for the microarray analysis this week. Your notebook entry should contain:
- The purpose: what was the purpose of your investigations?
- Your methods: what did you actually do? Give a step by step account.
- There should be enough detail provided so that you or another person could re-do it based solely on your notebook.
- You may copy protocol instructions on your page and modify them as to what you actually did, as long as you provide appropriate attribution.
- Take advantage of the electronic nature of the notebook by providing screenshots, links to web pages, links to data, etc.
- Your results: the answers to the questions in the protocol, plus any other results you gathered.
- Usually it makes sense to embed your answers to the questions/results in the "methods" in the order in which you obtained them.
- A scientific conclusion: what was your main finding for today's project? Did you fulfill the purpose? Why or why not?
Microarray Data Analysis
Before you begin: Turn on File Extensions
- The Windows 7 operating systems defaults to hiding file extensions. To turn them back on, do the following:
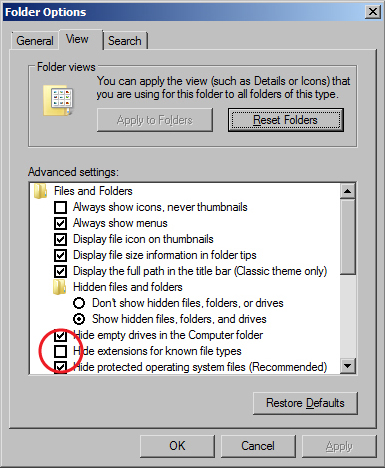
Folder Options window - Go to the Start menu and select "Control Panel".
- In the window that appears, search for "Folder Options" in the search field in the upper right hand corner.
- Click on "Folder Options" in the main window.
- When the Folder Options window appears, click on the View tab.
- Uncheck the box for "Hide extensions for known file types".
- Click the OK button.
- The computers in Seaver 120 are are set to erase all custom user settings and restore the defaults once they have been restarted, so if you have done this previously, you might have to do it again.

Experimental Design
Recall from last week's lecture, there were five timepoints in the experiment that were competitively hybridized with the t0 sample:
- t15 (cold shock)
- t30 (cold shock)
- t60 (cold shock)
- t90 (recovery)
- t120 (recovery)
Each timepoint has 3-5 independent replicates.
Note that the Δswi4 strain does not have a t15 timepoint and begins with t30.
Within-strain ANOVA
The purpose of the within-stain ANOVA test is to determine if any genes had a gene expression change that was significantly different than zero at any timepoint.
- Create a new worksheet, naming it either "(STRAIN)_ANOVA" as appropriate. For example, you might call yours "wt_ANOVA" or "dHAP4_ANOVA"
- Copy the first three columns containing the "Master_Index", "ID", and "Standard_Name" from the "Master_Sheet" worksheet for your strain and paste it into your new worksheet. Copy the columns containing the data for your strain and paste it into your new worksheet.
- At the top of the first column to the right of your data, create five column headers of the form (STRAIN)_AvgLogFC_(TIME) where (STRAIN) is your strain designation and (TIME) is 15, 30, etc.
- In the cell below the (STRAIN)_AvgLogFC_t15 header, type
=AVERAGE( - Then highlight all the data in row 2 associated with (STRAIN) and t15, press the closing paren key (shift 0),and press the "enter" key.
- This cell now contains the average of the log fold change data from the first gene at t=15 minutes.
- Click on this cell and position your cursor at the bottom right corner. You should see your cursor change to a thin black plus sign (not a chubby white one). When it does, double click, and the formula will magically be copied to the entire column of 6188 other genes.
- Repeat steps (4) through (8) with the t30, t60, t90, and the t120 data.
- Now in the first empty column to the right of the (STRAIN)_AvgLogFC_t120 calculation, create the column header (STRAIN)_ss_HO.
- In the first cell below this header, type
=SUMSQ( - Highlight all the LogFC data in row 2 for your (STRAIN) (but not the AvgLogFC), press the closing paren key (shift 0),and press the "enter" key.
- In the next empty column to the right of (STRAIN)_ss_HO, create the column headers (STRAIN)_ss_(TIME) as in (3).
- Make a note of how many data points you have at each time point for your strain. For most of the strains, it will be 4, but for dHAP4 t90 or t120, it will be "3", and for the wild type it will be "4" or "5". Count carefully. Also, make a note of the total number of data points. Again, for most strains, this will be 20, but for example, dHAP4, this number will be 18, and for wt it should be 23 (double-check).
- In the first cell below the header (STRAIN)_ss_t15, type
=SUMSQ(<range of cells for logFC_t15>)-<number of data points>*<AvgLogFC_t15>^2and hit enter.- The phrase <range of cells for logFC_t15> should be replaced by the data range associated with t15.
- The phrase <number of data points> should be replaced by the number of data points for that timepoint (either 3, 4, or 5).
- The phrase <AvgLogFC_t15> should be replaced by the cell number in which you computed the AvgLogFC for t15, and the "^2" squares that value.
- Upon completion of this single computation, use the Step (7) trick to copy the formula throughout the column.
- Repeat this computation for the t30 through t120 data points. Again, be sure to get the data for each time point, type the right number of data points, and get the average from the appropriate cell for each time point, and copy the formula to the whole column for each computation.
- In the first column to the right of (STRAIN)_ss_t120, create the column header (STRAIN)_SS_full.
- In the first row below this header, type
=sum(<range of cells containing "ss" for each timepoint>)and hit enter. - In the next two columns to the right, create the headers (STRAIN)_Fstat and (STRAIN)_p-value.
- Recall the number of data points from (13): call that total n.
- In the first cell of the (STRAIN)_Fstat column, type
=((n-5)/5)*(<(STRAIN)_ss_HO>-<(STRAIN)_SS_full>)/<(STRAIN)_SS_full>and hit enter.- Don't actually type the n but instead use the number from (13). Also note that "5" is the number of timepoints and the dSWI4 strain has 4 timepoints (it is missing t15).
- Replace the phrase (STRAIN)_ss_HO with the cell designation.
- Replace the phrase <(STRAIN)_SS_full> with the cell designation.
- Copy to the whole column.
- In the first cell below the (STRAIN)_p-value header, type
=FDIST(<(STRAIN)_Fstat>,5,n-5)replacing the phrase <(STRAIN)_Fstat> with the cell designation and the "n" as in (13) with the number of data points total. (Again, note that the number of timepoints is actually "4" for the dSWI4 strain). Copy to the whole column. - Before we move on to the next step, we will perform a quick sanity check to see if we did all of these computations correctly.
- Click on cell A1 and click on the Data tab. Select the Filter icon (looks like a funnel). Little drop-down arrows should appear at the top of each column. This will enable us to filter the data according to criteria we set.
- Click on the drop-down arrow on your (STRAIN)_p-value column. Select "Number Filters". In the window that appears, set a criterion that will filter your data so that the p value has to be less than 0.05.
- Excel will now only display the rows that correspond to data meeting that filtering criterion. A number will appear in the lower left hand corner of the window giving you the number of rows that meet that criterion. We will check our results with each other to make sure that the computations were performed correctly.
Calculate the Bonferroni and p value Correction
- Now we will perform adjustments to the p value to correct for the multiple testing problem. Label the next two columns to the right with the same label, (STRAIN)_Bonferroni_p-value.
- Type the equation
=<(STRAIN)_p-value>*6189, Upon completion of this single computation, use the Step (10) trick to copy the formula throughout the column. - Replace any corrected p value that is greater than 1 by the number 1 by typing the following formula into the first cell below the second (STRAIN)_Bonferroni_p-value header:
=IF(r2>1,1,r2). Use the Step (10) trick to copy the formula throughout the column.
Calculate the Benjamini & Hochberg p value Correction
- Insert a new worksheet named "(STRAIN)_ANOVA_B-H".
- Copy and paste the "Master_Index", "ID", and "Standard_Name" columns from your previous worksheet into the first two columns of the new worksheet.
- For the following, use Paste special > Paste values. Copy your unadjusted p values from your ANOVA worksheet and paste it into Column D.
- Select all of columns A, B, C, and D. Sort by ascending values on Column D. Click the sort button from A to Z on the toolbar, in the window that appears, sort by column D, smallest to largest.
- Type the header "Rank" in cell E1. We will create a series of numbers in ascending order from 1 to 6189 in this column. This is the p value rank, smallest to largest. Type "1" into cell E2 and "2" into cell E3. Select both cells E2 and E3. Double-click on the plus sign on the lower right-hand corner of your selection to fill the column with a series of numbers from 1 to 6189.
- Now you can calculate the Benjamini and Hochberg p value correction. Type (STRAIN)_B-H_p-value in cell F1. Type the following formula in cell F2:
=(D2*6189)/E2and press enter. Copy that equation to the entire column. - Type "STRAIN_B-H_p-value" into cell G1.
- Type the following formula into cell G2:
=IF(F2>1,1,F2)and press enter. Copy that equation to the entire column. - Select columns A through G. Now sort them by your Master_Index in Column A in ascending order.
- Copy column G and use Paste special > Paste values to paste it into the next column on the right of your ANOVA sheet.
Sanity Check: Number of genes significantly changed
Before we move on to further analysis of the data, we want to perform a more extensive sanity check to make sure that we performed our data analysis correctly. We are going to find out the number of genes that are significantly changed at various p value cut-offs.
- Go to your (STRAIN)_ANOVA worksheet.
- Select row 1 (the row with your column headers) and select the menu item Data > Filter > Autofilter (The funnel icon on the Data tab). Little drop-down arrows should appear at the top of each column. This will enable us to filter the data according to criteria we set.
- Click on the drop-down arrow for the unadjusted p value. Set a criterion that will filter your data so that the p value has to be less than 0.05.
- How many genes have p < 0.05? and what is the percentage (out of 6189)?
- How many genes have p < 0.01? and what is the percentage (out of 6189)?
- How many genes have p < 0.001? and what is the percentage (out of 6189)?
- How many genes have p < 0.0001? and what is the percentage (out of 6189)?
- When we use a p value cut-off of p < 0.05, what we are saying is that you would have seen a gene expression change that deviates this far from zero by chance less than 5% of the time.
- We have just performed 6189 hypothesis tests. Another way to state what we are seeing with p < 0.05 is that we would expect to see this a gene expression change for at least one of the timepoints by chance in about 5% of our tests, or 309 times. Since we have more than 309 genes that pass this cut off, we know that some genes are significantly changed. However, we don't know which ones. To apply a more stringent criterion to our p values, we performed the Bonferroni and Benjamini and Hochberg corrections to these unadjusted p values. The Bonferroni correction is very stringent. The Benjamini-Hochberg correction is less stringent. To see this relationship, filter your data to determine the following:
- How many genes are p < 0.05 for the Bonferroni-corrected p value? and what is the percentage (out of 6189)?
- How many genes are p < 0.05 for the Benjamini and Hochberg-corrected p value? and what is the percentage (out of 6189)?
- In summary, the p value cut-off should not be thought of as some magical number at which data becomes "significant". Instead, it is a moveable confidence level. If we want to be very confident of our data, use a small p value cut-off. If we are OK with being less confident about a gene expression change and want to include more genes in our analysis, we can use a larger p value cut-off.
- Comparing results with known data: the expression of the gene NSR1 (ID: YGR159C)is known to be induced by cold shock. Find NSR1 in your dataset. What is its unadjusted, Bonferroni-corrected, and B-H-corrected p values? What is its average Log fold change at each of the timepoints in the experiment? Note that the average Log fold change is what we called "STRAIN)_AvgLogFC_(TIME)" in step 3 of the ANOVA analysis.
- We will compare the numbers we get between the wild type strain and the other strains studied, organized as a table. Use this sample PowerPoint slide to see how your table should be formatted.
Clustering and GO Term Enrichment with stem
- Prepare your microarray data file for loading into STEM.
- Insert a new worksheet into your Excel workbook, and name it "(STRAIN)_stem".
- Select all of the data from your "(STRAIN)_ANOVA" worksheet and Paste special > paste values into your "(STRAIN)_stem" worksheet.
- Your leftmost column should have the column header "Master_Index". Rename this column to "SPOT". Column B should be named "ID". Rename this column to "Gene Symbol". Delete the column named "Standard_Name".
- Filter the data on the B-H corrected p value to be > 0.05 (that's greater than in this case).
- Once the data has been filtered, select all of the rows (except for your header row) and delete the rows by right-clicking and choosing "Delete Row" from the context menu. Undo the filter. This ensures that we will cluster only the genes with a "significant" change in expression and not the noise.
- Delete all of the data columns EXCEPT for the Average Log Fold change columns for each timepoint (for example, wt_AvgLogFC_t15, etc.).
- Rename the data columns with just the time and units (for example, 15m, 30m, etc.).
- Save your work. Then use Save As to save this spreadsheet as Text (Tab-delimited) (*.txt). Click OK to the warnings and close your file.
- Note that you should turn on the file extensions if you have not already done so.
- Now download and extract the STEM software. Click here to go to the STEM web site.
- Click on the download link, register, and download the
stem.zipfile to your Desktop. - Unzip the file. In Seaver 120, you can right click on the file icon and select the menu item 7-zip > Extract Here.
- This will create a folder called
stem. Inside the folder, double-click on thestem.jarto launch the STEM program.
- Click on the download link, register, and download the
- Running STEM
- In section 1 (Expression Data Info) of the the main STEM interface window, click on the Browse... button to navigate to and select your file.
- Click on the radio button No normalization/add 0.
- Check the box next to Spot IDs included in the data file.
- In section 2 (Gene Info) of the main STEM interface window, select Saccharomyces cerevisiae (SGD), from the drop-down menu for Gene Annotation Source. Select No cross references, from the Cross Reference Source drop-down menu. Select No Gene Locations from the Gene Location Source drop-down menu.
- In section 3 (Options) of the main STEM interface window, make sure that the Clustering Method says "STEM Clustering Method" and do not change the defaults for Maximum Number of Model Profiles or Maximum Unit Change in Model Profiles between Time Points.
- In section 4 (Execute) click on the yellow Execute button to run STEM.
- In section 1 (Expression Data Info) of the the main STEM interface window, click on the Browse... button to navigate to and select your file.
- Viewing and Saving STEM Results
- A new window will open called "All STEM Profiles (1)". Each box corresponds to a model expression profile. Colored profiles have a statistically significant number of genes assigned; they are arranged in order from most to least significant p value. Profiles with the same color belong to the same cluster of profiles. The number in each box is simply an ID number for the profile.
- Click on the button that says "Interface Options...". At the bottom of the Interface Options window that appears below where it says "X-axis scale should be:", click on the radio button that says "Based on real time". Then close the Interface Options window.
- Take a screenshot of this window (on a PC, simultaneously press the
AltandPrintScreenbuttons to save the view in the active window to the clipboard) and paste it into a PowerPoint presentation to save your figures.
- Click on each of the SIGNIFICANT profiles (the colored ones) to open a window showing a more detailed plot containing all of the genes in that profile.
- Take a screenshot of each of the individual profile windows and save the images in your PowerPoint presentation.
- At the bottom of each profile window, there are two yellow buttons "Profile Gene Table" and "Profile GO Table". For each of the profiles, click on the "Profile Gene Table" button to see the list of genes belonging to the profile. In the window that appears, click on the "Save Table" button and save the file to your desktop. Make your filename descriptive of the contents, e.g. "wt_profile#_genelist.txt", where you replace the number symbol with the actual profile number.
- Upload these files to OpenWetWare and link to them on your individual journal page. (Note that it will be easier to zip all the files together and upload them as one file).
- For each of the significant profiles, click on the "Profile GO Table" to see the list of Gene Ontology terms belonging to the profile. In the window that appears, click on the "Save Table" button and save the file to your desktop. Make your filename descriptive of the contents, e.g. "wt_profile#_GOlist.txt", where you use "wt", "dGLN3", etc. to indicate the dataset and where you replace the number symbol with the actual profile number. At this point you have saved all of the primary data from the STEM software and it's time to interpret the results!
- Upload these files to OpenWetWare and link to them on your individual journal page. (Note that it will be easier to zip all the files together and upload them as one file).
- A new window will open called "All STEM Profiles (1)". Each box corresponds to a model expression profile. Colored profiles have a statistically significant number of genes assigned; they are arranged in order from most to least significant p value. Profiles with the same color belong to the same cluster of profiles. The number in each box is simply an ID number for the profile.
- Analyzing and Interpreting STEM Results
- Select one of the profiles you saved in the previous step for further intepretation of the data. I suggest that you choose one that has a pattern of up- or down-regulated genes at the cold shock timepoints. Each member of your group should choose a different profile. Answer the following:
- Why did you select this profile? In other words, why was it interesting to you?
- How many genes belong to this profile?
- How many genes were expected to belong to this profile?
- What is the p value for the enrichment of genes in this profile? Bear in mind that we just finished computing p values to determine whether each individual gene had a significant change in gene expression at each time point. This p value determines whether the number of genes that show this particular expression profile across the time points is significantly more than expected.
- Open the GO list file you saved for this profile in Excel. This list shows all of the Gene Ontology terms that are associated with genes that fit this profile. Select the third row and then choose from the menu Data > Filter > Autofilter. Filter on the "p-value" column to show only GO terms that have a p value of < 0.05. How many GO terms are associated with this profile at p < 0.05? The GO list also has a column called "Corrected p-value". This correction is needed because the software has performed thousands of significance tests. Filter on the "Corrected p-value" column to show only GO terms that have a corrected p value of < 0.05. How many GO terms are associated with this profile with a corrected p value < 0.05?
- Select 6 Gene Ontology terms from your filtered list (either p < 0.05 or corrected p < 0.05).
- Each member of the group will be reporting on his or her own cluster in your presentation next week. You should take care to choose terms that are the most significant, but that are also not too redundant. For example, "RNA metabolism" and "RNA biosynthesis" are redundant with each other because they mean almost the same thing.
- Note whether the same GO terms are showing up in multiple clusters.
- Look up the definitions for each of the terms at http://geneontology.org. In your final presentation, you will discuss the biological interpretation of these GO terms. In other words, why does the cell react to cold shock by changing the expression of genes associated with these GO terms? Also, what does this have to do with the transcription factor being deleted (for the Δgln3 and Δswi4 groups)?
- To easily look up the definitions, go to http://geneontology.org.
- Copy and paste the GO ID (e.g. GO:0044848) into the search field at center top of the page called "Search GO Data".
- In the results page, click on the button that says "Link to detailed information about <term>, in this case "biological phase"".
- The definition will be on the next results page, e.g. here.
- Each member of the group will be reporting on his or her own cluster in your presentation next week. You should take care to choose terms that are the most significant, but that are also not too redundant. For example, "RNA metabolism" and "RNA biosynthesis" are redundant with each other because they mean almost the same thing.
- Select one of the profiles you saved in the previous step for further intepretation of the data. I suggest that you choose one that has a pattern of up- or down-regulated genes at the cold shock timepoints. Each member of your group should choose a different profile. Answer the following:
Data and Files
- Files left on the Desktop or My Documents or Downloads folders on the Seaver 120 computers will be deleted upon restart of the computers. Files stored on the
T:drive will be saved. However, it is not a good idea to trust that they will be there when you next use the computer. - Thus, it is a critical skill for data and computer literacy to back-up your data and files in at least two ways:
- Upload the files to this wiki.
- Upload the files to Box.
- Back them up on your personal flash drive.
- References to data and files should be made within the methods and results section of your notebook, listed above.
- In addition to these inline links, create a "Data and Files" section of your notebook to make a list of the files generated in this exercise. The files you will need to save and link to in order to document your work are as follows:
- Your Excel workbook containing the statistical analysis
- The input file for stem
- Zipped _genelist.txt files from stem
- Zipped _GOlist.txt files from stem
- Your statistics data table and screenshots of the stem results should be uploaded as a PowerPoint presentation. Note that this is different than your group presentation.
Final Group Presentation
- As noted last week, your final group presentation will combine elements of a journal club and a research presentation. Besides the journal club slides that you improved from last week, you will include the following from this week's work:
- Add information about your research results to the Outline slide
- The experimental design of the Dahlquist Lab microarray experiment and which strain you studied. Compare this to the strain and experiment conducted in the journal article you read.
- A table that lists the number of genes with significant changes in expression for the within-strain ANOVA at the p value cut-offs of < 0.05, < 0.01, < 0.001, and Bonferroni-corrected p < 0.05.
- Screenshots of the overall stem results for your strain and the four plots for the clusters that your group members individually analyzed. For each of the plots, include the 6 relevant GO terms that you selected to interpret for each cluster, along with the summary statistics from the _GOlist.txt file.
- Discussion and interpretation of your results, in comparison to the original journal club paper.
- Upload your slides to the OpenWetWare wiki by the Week 15 journal assignment deadline. Each member of the group should be linking to the same presentation. You may make changes to your slides in advance of your presentation, but you will be graded on what you upload by the journal deadline.
- Use these Presentation Guidelines when preparing your PowerPoint slides.
- Your presentation will also be graded on the following Presentation Rubric.
Academic Honesty
As discussed in class, each weekly individual journal assignment needs to conclude with an Acknowledgments and References section.
Acknowledgments
In this section, you need to acknowledge anyone who assisted you with your assignment, either in person, electronically, or even anonymously without their knowledge (see below).
- You must acknowledge your homework partner or team members with whom you worked, giving details of the nature of the collaboration. An appropriate statement could be (but is not limited to) the following:
- I worked with my homework partner (give name and link name to their user page) in class. We met face-to-face one time outside of class. We texted/e-mailed/chatted online three times. We worked on the <details> portion of the assignment together.
- Sign this statement with your wiki signature.
- I worked with my homework partner (give name and link name to their user page) in class. We met face-to-face one time outside of class. We texted/e-mailed/chatted online three times. We worked on the <details> portion of the assignment together.
- Acknowledge anyone else you worked with who was not your assigned partner. This could be Dr. Dahlquist (for example, via office hours), the TA, other students in the class, or even other students or faculty outside of the class.
- If you copied
wiki syntaxor a particular style from another wiki page, acknowledge that here. Provide the user name of the original page, if possible, and provide a link to the page from which you copied the syntax or style. If you need to reference content, use your References section (see below). - You must also include this statement unless otherwise noted: "While I worked with the people noted above, this individual journal entry was completed by me and not copied from another source."
References
- In this section, you need to provide properly formatted citations to any content that was not entirely of your own devising. This includes, but is not limited to:
- data
- facts
- images
- documents, including the scientific literature
- The references in this section should be accompanied by in text citations on your page that refer to these references.
- The references should be formatted according to the APA guidelines.
- For more detailed guidelines, please see the document Guidelines for Literature Citations in a Scientific Paper that you were given on the first day of class.
Shared Journal Assignment
- Store your journal entry in the shared BIOL368/F16:Class Journal Week 15 page. If this page does not exist yet, go ahead and create it.
- Link to the shared journal entry from your user page; this should be part of your template.
- Link the shared journal page to this assignment page.
- Sign your portion of the journal with the standard wiki signature shortcut (
~~~~). - Add the "BIOL368/F16" category to the end of the wiki page (if someone has not already done so).
Reflection
For your last reflection you will reflect on this entire semester:
- What is the most important thing that you learned this semester in this class?
- With your head (biological or bioinformatics principles)
- With your heart (personal qualities and teamwork qualities that make things work or not work)?
- With your hands (technical skills)?
- What will you take away from this class that you will still use a year from now?
