Moneil5 Week 12
Helpful Links
Assignment Pages:
- Week 1 Assignment
- Week 2 Assignment
- Week 3 Assignment
- Week 4 Assignment
- Week 5 Assignment
- Week 6 Assignment
- Week 7 Assignment
- Week 9 Assignment
- Week 10 Assignment
- Week 11 Assignment
- Week 12 Assignment
- Week 14/15 Assignment
Personal Journal Entries:
- Week 1
- Week 2
- Week 3
- Week 4
- Week 5
- Week 6
- Week 7
- Week 7 Re-visited
- Week 9
- Week 10
- Week 11
- Week 12
- Week 14/15
Shared Journal Entries:
Purpose
The purpose of this assignment is to continue in researching potential gene regulatory networks based on our given yeast strain. We are meant to generate a potential graph by the end of this assignment. The purpose is also to understand how the model uses each data sheet in the excel input files, and how those sheets are created.
Workflow/Methods
Using Cluster 45 from Week 11 Assignment
Using YEASTRACT to Infer which Transcription Factors Regulate a Cluster of Genes
In the previous analysis using STEM, we found a number of gene expression profiles (aka clusters) which grouped genes based on similarity of gene expression changes over time. The implication is that these genes share the same expression pattern because they are regulated by the same (or the same set) of transcription factors. We will explore this using the YEASTRACT database.
- Open the gene list in Excel for the one of the significant profiles from your stem analysis. Choose a cluster with a clear cold shock/recovery up/down or down/up pattern. You should also choose one of the largest clusters.
- Copy the list of gene IDs onto your clipboard.
- Launch a web browser and go to the YEASTRACT database.
- On the left panel of the window, click on the link to Rank by TF.
- Paste your list of genes from your cluster into the box labeled ORFs/Genes.
- Check the box for Check for all TFs.
- Accept the defaults for the Regulations Filter (Documented, DNA binding plus expression evidence)
- Do not apply a filter for "Filter Documented Regulations by environmental condition".
- Rank genes by TF using: The % of genes in the list and in YEASTRACT regulated by each TF.
- Click the Search button.
- Answer the following questions:
- In the results window that appears, the p values colored green are considered "significant", the ones colored yellow are considered "borderline significant" and the ones colored pink are considered "not significant". How many transcription factors are green or "significant"?
- 18 transcription factors are significant
- Copy the table of results from the web page and paste it into a new Excel workbook to preserve the results
- Are GLN3 or HAP4 on the list? If so, what is their "% in user set", "% in YEASTRACT", and "p value".
- GLN3 and HAP4 are not on the list for this cluster
- For the mathematical model that we will build, we need to define a gene regulatory network of transcription factors that regulate other transcription factors. We can use YEASTRACT to assist us with creating the network. We want to generate a network with approximately 15-30 transcription factors in it.
- You need to select from this list of "significant" transcription factors, which ones you will use to run the model. You will use these transcription factors and add GLN3 and HAP4 if they are not in your list. Explain in your electronic notebook how you decided on which transcription factors to include. Record the list and your justification in your electronic lab notebook.
- I only had 18 genes pop up in my list, and didn't have GLN3 or HAP4 so I stuck with my original list, plus GLn3 and HAP4 to plug into the matrix generator
- Go back to the YEASTRACT database and follow the link to Generate Regulation Matrix.
- Copy and paste the list of transcription factors you identified (plus HAP4 and GLN3) into both the "Transcription factors" field and the "Target ORF/Genes" field.
- We are going to use the "Regulations Filter" options of "Documented", "Only DNA binding evidence"
- Click the "Generate" button.
- In the results window that appears, click on the link to the "Regulation matrix (Semicolon Separated Values (CSV) file)" that appears and save it to your Desktop. Rename this file with a meaningful name so that you can distinguish it from the other files you will generate.
- Ran file through GRNsight to determine which we connected to other genes. Eliminated NDT80 because it was connected to nothing. Ran file through Gephi using this protocol (insert Gephi protocol here). Deleted transcription factors that had an in degree of 0 according to Gephi, because in previous research it was found those nodes not regulated by anything performed more poorly in the model. End network has 14 nodes, 17 edges
- You need to select from this list of "significant" transcription factors, which ones you will use to run the model. You will use these transcription factors and add GLN3 and HAP4 if they are not in your list. Explain in your electronic notebook how you decided on which transcription factors to include. Record the list and your justification in your electronic lab notebook.
Visualizing Your Gene Regulatory Networks with GRNsight
We will analyze the regulatory matrix files you generated above in Microsoft Excel and visualize them using GRNsight to determine which one will be appropriate to pursue further in the modeling.
- First we need to properly format the output files from YEASTRACT. You will repeat these steps for each of the three files you generated above.
- Open the file in Excel. It will not open properly in Excel because a semicolon was used as the column delimiter instead of a comma. To fix this, Select the entire Column A. Then go to the "Data" tab and select "Text to columns". In the Wizard that appears, select "Delimited" and click "Next". In the next window, select "Semicolon", and click "Next". In the next window, leave the data format at "General", and click "Finish". This should now look like a table with the names of the transcription factors across the top and down the first column and all of the zeros and ones distributed throughout the rows and columns. This is called an "adjacency matrix." If there is a "1" in the cell, that means there is a connection between the trancription factor in that row with that column.
- Save this file in Microsoft Excel workbook format (.xlsx).
- Check to see that all of the transcription factors in the matrix are connected to at least one of the other transcription factors by making sure that there is at least one "1" in a row or column for that transcription factor. If a factor is not connected to any other factor, delete its row and column from the matrix. Make sure that you still have somewhere between 15 and 30 transcription factors in your network after this pruning.
- Only delete the transcription factor if there are all zeros in its column AND all zeros in its row. You may find visualizing the matrix in GRNsight (below) can help you find these easily.
- For this adjacency matrix to be usable in GRNmap (the modeling software) and GRNsight (the visualization software), we need to transpose the matrix. Insert a new worksheet into your Excel file and name it "network". Go back to the previous sheet and select the entire matrix and copy it. Go to you new worksheet and click on the A1 cell in the upper left. Select "Paste special" from the "Home" tab. In the window that appears, check the box for "Transpose". This will paste your data with the columns transposed to rows and vice versa. This is necessary because we want the transcription factors that are the "regulatORS" across the top and the "regulatEES" along the side.
- The labels for the genes in the columns and rows need to match. Thus, delete the "p" from each of the gene names in the columns. Adjust the case of the labels to make them all upper case.
- In cell A1, copy and paste the text "rows genes affected/cols genes controlling".
- Finally, for ease of working with the adjacency matrix in Excel, we want to alphabatize the gene labels both across the top and side.
- Select the area of the entire adjacency matrix.
- Click the Data tab and click the custom sort button.
- Sort Column A alphabetically, being sure to exclude the header row.
- Now sort row 1 from left to right, excluding cell A1. In the Custom Sort window, click on the options button and select sort left to right, excluding column 1.
- Name the worksheet containing your organized adjacency matrix "network" and Save.
- Now we will visualize what these gene regulatory networks look like with the GRNsight software.
- Go to the GRNsight home page.
- Select the menu item File > Open and select the regulation matrix .xlsx file that has the "network" worksheet in it that you formatted above. If the file has been formatted properly, GRNsight should automatically create a graph of your network. Move the nodes (genes) around until you get a layout that you like and take a screenshot of the results. Paste it into your PowerPoint presentation.
- Now we will look at some of the network properties. Again, repeat these steps for each of the three gene regulatory matrices you generated above. See this file for an example of how to do the following instructions.
- Create a new worksheet and call it "degree". Copy and paste your adjacency matrix from the "network" sheet into this new worksheet.
- In the first empty cell in column A, type "Out-degree". In the cell to the right of that in Column B, type the equation
=SUM(and select the range of cells in column B that has 1's and 0's in it, close the parentheses, and press Enter. This quantity is the number of genes that the transcription factor in that column is controlling, or the out-degree. Copy and paste that equation across all of the columns. - In Cell 1 of the first empty column to the right of the adjacency matrix, type "In-degree". In Cell 2 of this column, type the equation
=SUM(and select the entire row of 1's and 0's, close the parentheses, and press Enter. This quantity is the number of transcription factors that regulate the gene in that row, or the in-degree. Copy and paste the equation down the entire column, including the row that contains the out-degree sums. - The number in the lower right-hand corner, the sum of sums, is the total number of edges in the adjacency matrix. We would like to see about 50 (40-60 or so) edges in the matrix. If the matrix is too dense, it will slow down the modeling program because it will be difficult to estimate the parameters in the model.
- We want to plot the degree distributions for each of your gene regulatory networks. In the "degree" worksheet, create three columns to the right called "Frequency", "In-degree total", and "Out-degree total". In the "Frequency" column, number sequentially from 1 to the largest degree number in your calculations above. In the "In-degree total" column, type the number of genes with that in-degree for each of the frequencies. In the "Out-degree total" column, type the number of genes with that out-degree for each of the frequencies.
- Select the "Frequency", "In-degree total", and "Out-degree total" columns. Go to the "Insert" tab and select the column chart type to insert a plot of the degree distribution. Copy and paste the charts for each gene regulatory matrix into your PowerPoint presentation.
Generating an Input Workbook for GRNmap
Now that you have identified the gene regulatory network that you want to model, the next step is to generate the input Excel workbook that you will run in the GRNmap modeling software.
Click here to download a sample workbook on which to base the one specific to your network and microarray data.
Note that when following the instructions below, you need to follow them precisely, to the letter, or GRNmap will return an error.
production_rates sheet
- This sheet contains initial guesses for the production rate parameters, P, for all genes in the network.
- Assuming that the system is in steady state with the relative expression of all genes equal to 1, (P/2) - lambda = 0, where lambda is the degradation rate, is a reasonable initial guess.
- The sheet should contain two columns (from left to right) entitled, "id", "production_rate".
- The id is an identifier that the user will use to identify a particular gene.
- The "production_rate" column should then contain the initial guesses for the P parameter as described above, rounded to four decimal places.
- The genes should be listed in the same order in all the sheets in the Excel workbook.
- While this is the first worksheet in the workbook (from left to right), it makes sense to create the degradation_rates sheet first (instructions below).
degradation_rates sheet
- This sheet contains degradation rates for all genes in the network, which are provided by the user.
- Currently, the Dahlquist Lab is using data based on published mRNA half-life data from Neymotin et al. (2006).
- We converted the half-life data values to the degradation rates by taking the natural log of the half-life and dividing by 2.
- The sheet should contain two columns (from left to right) entitled "id", and "degradation_rate".
- The id is an identifier that the user will use to identify a particular gene.
- The "degradation_rate" column should then contain the absolute value of the degradation rate for the corresponding gene as described above, rounded to four decimal places.
- To obtain these values, download the file: Neymotin_2014_RNA_degradation_rates_processed.xlsx.
- From the worksheet called "degradation_rate_Harbison202", locate the rounded degradation rate value (from column F) and copy and paste it into your input workbook.
- If the value is missing for a gene, check the worksheet "degradation_rate_all". If it is not there either, substitute the median value of -0.0990 for the missing degradation rates.
- The genes should be listed in the same order in all the sheets in the Excel workbook.
Expression Data Sheets for Individual Yeast Strains
- Expression data can be provided for either a single strain or multiple strains of yeast (for example, the wild type strain and a transcription factor deletion strain).
- Each strain will have its own sheet in the workbook.
- Each sheet should be given a unique name that follows the convention "STRAIN_log2_expression", where the word "STRAIN" is replaced by the strain designation, which will appear in the optimization_diagnostics sheet. The sheet should have the following columns in this order:
- "id": list of all genes. The genes should be listed in the same order in all the sheets in the Excel workbook.
- The next series of columns should contain the expression data for each gene at a given timepoint given as log2 ratios (log2 fold changes). The column header should be the time at which the data were collected, without any units. For example, the 15 minute timepoint would have a column header "15" and the 30 minute timepoint would have the column header "30". GRNmap supports replicate data for each of the timepoints. Replicate data for the same timepoint should be in columns immediately next to each other and have the same column headers. For example, three replicates of the 15 minute timepoint would have "15", "15", "15" as the column headers.
- If data are provided for multiple strains, each strain should have data for the same timepoints.
- Everyone in the class will have at least one expression worksheet called "wt_log2_expression".
- If your network includes GLN3 and/or HAP4, then include expression worksheets, called "dgln3_log2_expression" and/or "dhap4_log2_expression".
- Include the data for the 15, 30, and 60 minute timepoints, but not the 90 or 120 minute timepoints.
- The data you will be using is the normalized log2 fold changes that you did the ANOVA analysis on. Remember, the data files are on Box.
- "BIOL398-05_Spring2017_master_microarray_data_wt.xlsx"
- "BIOL398-05_Spring2017_master_microarray_data_dGLN3.xlsx"
- "BIOL398-05_Spring2017_master_microarray_data_dHAP4.xlsx"
- It is tedious to copy and paste all of the data by hand, so in class, Dr. Dahlquist will demonstrate how to execute a query in Microsoft Access to do it for you.
- You will note that in the sample input workbook, there are some cells in the expression sheets highlighted in yellow. This is because those cells had missing data in them. GRNmap will return an error if it encounters missing values, so we need to fill those cells.
- When you see cells with missing data in them, highlight the cell yellow.
- Then write a formula that computes the average of the log2 fold change for that gene and timepoint.
- Then copy the contents of that cell, and "Paste special > paste values" into the cell to remove the formula.
network sheet
- This sheet contains an adjacency matrix representation of the gene regulatory network.
- The columns correspond to the transcription factors and the rows correspond to the target genes controlled by those transcription factors.
- A “1” means there is an edge connecting them and a “0” means that there is no edge connecting them.
- The upper-left cell (A1) should contain the text “cols regulators/rows targets”. This text is there as a reminder of the direction of the regulatory relationships specified by the adjacency matrix.
- The rest of row 1 should contain the names of the transcription factors that are controlling the other genes in the network, one transcription factor name per column.
- The rest of column A should contain the names of the target genes that are being controlled by the transcription factors heading each of the columns in the matrix, one target gene name per row.
- The transcription factor names should correspond to the "id" in the other sheets in the workbook. They should be capitalized the same way and occur in the same order along the top and side of the matrix. The matrix needs to be symmetric, i.e., the same transcription factors should appear along the top and left side of the matrix. The genes should be listed in the same order in all the sheets in the Excel workbook.
- Each cell in the matrix should then contain a zero (0) if there is no regulatory relationship between those two transcription factors, or a one (1) if there is a regulatory relationship between them. Again, the columns correspond to the transcription factors and the rows correspond to the target genes controlled by those transcription factors.
- The network you derived from the YEASTRACT database in the last class can be copied and pasted into this sheet directly. You may need to edit the contents of cell A1, but the rest should be good to go (especially if you previewed it in GRNsight).
network_weights sheet
- These are the initial guesses for the estimation of the weight parameters, w.
- Since these weights are initial guesses which will be optimized by GRNmap, the content of this sheet can be identical to the "network" sheet.
optimization_parameters sheet
The optimization_parameters sheet should have two columns (from left to right) entitled, "optimization_parameter" and "value".
You should copy this worksheet from the sample workbook provided. The only row that you need to modify is row 15, "Strain". Include just the strain designations for which you have a corresponding STRAIN_log2_expression sheet. Everyone will have "wt" and some will have either or both "dgln3" and/or "dhap4".
This is an explanation of what the optimization_parameters mean.
- alpha: Penalty term weighting (from the L-curve analysis)
- kk_max: Number of times to re-run the optimization loop. In some cases re-starting the optimization loop can improve performance of the estimation.
- MaxIter: Number of times MATLAB iterates through the optimization scheme. If this is set too low, MATLAB will stop before the parameters are optimized.
- TolFun: How different two least squares evaluations should be before the program determines that it is not making any improvement
- MaxFunEval: maximum number of times the program will evaluate the least squares cost
- TolX: How close successive least squares cost evaluations should be before the program determines that it is not making any improvement.
- production_function: = Sigmoid (case-insensitive) if sigmoidal model, =MM (case-insensitive) if Michaelis-Menten model
- L_curve: =0 if an L-curve analysis should NOT be run or =1 if an L-curve analysis SHOULD be run. The L-curve analysis will automatically run sequential rounds of estimation for an array of fixed alpha values (0.8, 0.5, 0.2, 0.1,0.08, 0.05,0.02,0.01, 0.008, 0.005, 0.002, 0.001, 0.0008, 0.0005, 0.0002, and 0.0001). GRNmap makes a copy of the user's selected input workbook and changes alpha to the first alpha in the list. The estimation runs and the resulting parameter values are used as the initial guesses for the next round of estimation with the next alpha value. This process repeats until all alpha values have been run. New input and output workbooks are generated for each alpha value, although currently, the graphs are only saved for the last run.
- estimate_params =1 if want to estimate parameters and =0 if the user wants to do just one forward run
- make_graphs =1 to output graphs; =0 to not output graphs
- fix_P =1 if the user does not want to estimate the production rate, P, parameter, just use the initial guess and never change; =0 to estimate
- fix_b =1 if the user does not want to estimate the b parameter, just use the initial guess and never change; =0 to estimate
- expression_timepoints: A row containing a list of the time points when the data was collected experimentally. Should correspond to the timepoint column headers in the STRAIN_log2_expression sheets.
- Strain: A row containing a list of all of the strains for which there is expression data in the workbook. Should correspond to the "STRAIN" portion of the names of the STRAIN_log2_expression sheets for each strain. Note that GRNmap will run the model for the wild type network (all genes present in the network) and for networks where the gene deleted from the designated STRAIN has been deleted from the network.
- simulation_timepoints: A row containing a list of the time points at which to evaluate the differential equations to generate the simulated data. This does not need to correspond to the actual measurement times, but should be in the same units (e.g. minutes).
threshold_b sheet
- These are the initial guesses for the estimation of the threshold_b parameters.
- There should be two columns.
- The left-most column should contain the header "id" and list the standard names for the genes in the model in the same order as in the other sheets.
- The second column should have the header "threshold_b" and should contain the initial guesses, typically all 0.
Once you have completed your input workbook, either upload and link to it on the OWW wiki, or share it on Box.
Results
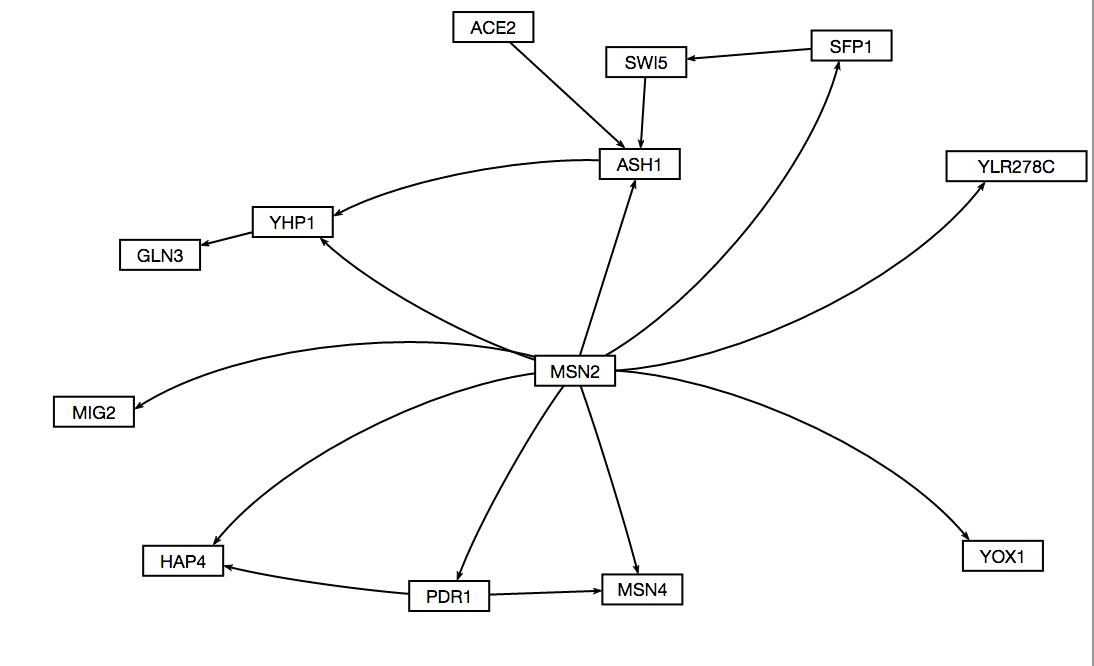
18 genes from Cluster 45 were found to be significant, and after pairing down nodes that didn't play much of a role in the network, what was left is a network consisting of 13 nodes and 16 edges.
Visualization
The following is a visual of the network created using GRNsight
GRNmap Input
The workbook generated for this assignment can be found here:
Conclusion
This week the purpose was achieved. A 13 node, 16 edge graph was generated from cluster 45 as a potential GRN for the cold shock response in yeast based on the wild type strain data. The purpose was met because the underlying methods used to create the input workbooks was learned, and a strong potential graph was generated as well.
References
Dahlquist, Kam D. (2017) BIOL398-05/S17:Week 12. Retrieved from http://www.openwetware.org/wiki/BIOL398-05/S17:Week_12 on 11 April 2017
Acknowledgments
- Assignment was completed using the Week 12 assignment protocol (pasted into the methods section of this entry). This assignment can be found at the page link in the references section.
- Except for what is noted above, this individual journal entry was completed by me and not copied from another source.
Margaret J. Oneil 23:28, 19 April 2017 (EDT)
