BIOL368/S19 Week 5
- Weekly Assignments
- Individual Journal Entries
- Fatimah Alghanem Journal Entry Week 2
- Fatimah Alghanem Journal Entry Week 3
- Fatimah Alghanem Journal Entry Week 4
- Fatimah Alghanem Journal Entry Week 5
- Fatimah Alghanem Journal Entry Week 6
- Fatimah Alghanem Journal Entry Week 7
- DrugComboDB Review
- Fatimah Alghanem Journal Entry Week 9
- Fatimah Alghanem Journal Entry Week 10
- Fatimah Alghanem Journal Entry Week 11
- The Mutants Research Project Week 12
- Fatimah Alghanem Journal Entry Week 14
- Class Journal
BIOL368/F20 BIOL368/F20:People
Purpose
The purpose of this assignment is to learn about different protein structures and be able to identify the different information that each structure gives. Also to use the NCBI Structure database to find details regarding specific protein structures.
Methods and results
- I visited UniProt Knowledgebase (UniProt KB) and searched using the keyword "SARS-CoV-2" in the main UniProt search field and got 1,616 results. Not All the results were viral proteins as there were some mouse proteins too.
- I used the entry with accession number "P0DTC2" which corresponds to the reference entry for the SARS-CoV-2 spike protein to find information about the protein and they were as follows:
- Spike proteins include S1, S2, and S2'.
- S1 binds to ACE2.
- I Opened the NCBI Structure database and chose Figure 1A SARS-CoV RBD (year 2002) complexed with human ACE2: 2AJF to work on.

- Figure 1. SARS-CoV RBD (year 2002) complexed with human ACE2
- In the figure above demonstrates a quaternary structure as there are two amino acid chains connected to one another.
- There are two domains in the figure above following that it's a quaternary structure with two proteins connected to one another.
- I clicked on the Style > Proteins menu and showed screenshots of the following styles:
- Cylinder and Plate
- C Alpha Trace
- Lines
- Ball and Stick
- Spheres





- I answered what's unique about each style
- Cylinder and plate: show the helicases clearly and it's easy to identify if it's an Alpha/ Beta Helix as the alpha helix is shown as cylinders.
- C Alpha Trace: easy to look at and not too complicated. However, it doesn't show much.
- Lines: this style shows everything as lines and it makes it easy to see the backbone but doesn't give much information in terms of protein structures.
- Ball and Stick: all the atoms are shown as balls making it easy to follow atoms order.
- Sphere: this shows a more realistic representation of the space that atoms take by each atom having its own sphere.
- In the “Spheres” view, I clicked on the Color menu and show screenshots of the following color schemes:
- spectrum
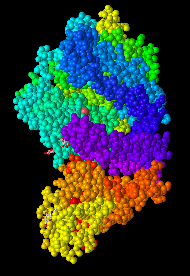
- This color scheme divides the structure based on color but I am not sure what it's dividing it too.
- Secondary
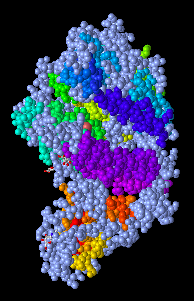
- This shows the secondary structure in the whole protein.
- Charge

- This color scheme identify the parts of the structure by charges. Amino acids with no charges are grey. Basic and acidic amino acids are represented by blue and red respectively. This is a good representation to demonstrate the amino acids that are in the structures.
- Atom

- This distinguishes between atoms by color, (N+: red), (O2: blue), (C: grey), (S: Yellow). This is a good way to visualize the atoms that are within the structure and what is creating it.




- The N-terminus and C-terminus of each polypeptide were found.

- The secondary structures in ACE2 were found to include: Beta sheet identified by the arrows, alpha helix identified by the rod shape, and random coil identified by not having a well-defined shape.
- The secondary structures in the spike protein were found to be: Beta sheet identified by the arrows and random coil by not having a well-defined shape.
- I looked at the side chain shown in Figure 4B 3SCK.
- I clicked in this link civet ACE2-spike protein structure to interact with the structure in iCn3D.
- Then I clicked on the Windows menu to “View Sequences & Annotations”
- In the new window that appears to the right, I clicked on the “Details” tab to show the actual amino acid sequences.
- 2 sets of ACE2-spike proteins were seen because of the way the proteins crystallized.
- I focused on the pink and tan chains and oriented them like is shown in Figure 4B
- In the sequence window I went to sequence “Protein 3SCK_A” (in pink) and select the following amino acids: T31, E35, E38, T82, and K353.
- The part of the ribbon that represents these amino acids were shown highlighted in yellow in the structure.
- I went to the Styles menu and selected Proteins > Ball and Stick
- Then I went to the color menu and select Atom
- The side chains were shown in the figure
- The same process was repeated for the tan spike protein sequence 3SCK_E for the following amino acids: T487, R479,G480, Y442
P472.
- Screenshots were then taken of both figures oriented like in Figure 4B:
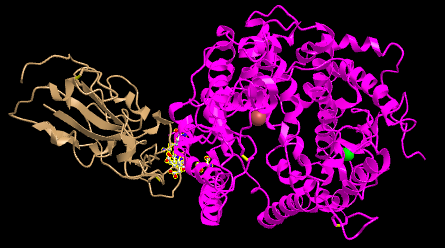
- Protein 3SCK_A
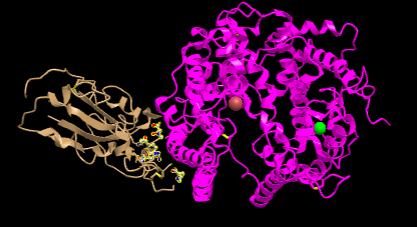
- Protein 3SCK_E


- The following questions were answered
- E35 on ACE2 makes an ionic bond with R479 on spike protein. Which of these is acidic and which is basic? How can you tell that from the image you created?
- E35 is the acidic one since it is Glutamic "acid" from the image E35 is demonstrated in red which means that it's acidic and R479 is demonstrated in blue showing that it's basic.
- T31 on ACE2 makes a hydrogen bond with Y442 on spike protein. Which side-chain classification do these two amino acids belong to (acidic, basic, uncharged polar, nonpolar)? How can you tell that from the image you created?
- Both Tyrosine and Threonine are uncharged polar and that's represented by the hydroxyl group in the picture.
- Dash lines for the bond was made following these steps:
- I went to the View and select H bonds & Interactions
- In part 1 of the window that appears, I unchecked “Contacts/Interactions” and left Hydrogen Bonds and Ionic Interaction checked
- In part 2 of the window, I selected the first set “3SCK_A” (pink)
- In part 3 of the window, I selected the second set “3SCK_E” (tan)
- In part 4 of the window, I clicked the button “3D Display”
- The dashed lines representing the ionic bonds and H-bonds between the two polypeptide chains and amino acids were seen as shown in the picture below:

Beginning your research project
- What question will you answer about sequence-->structure-->function relationships in the spike and/or ACE2 protein?
- The data presented in Wan et al (2020) suggests that a single mutation in N501T which corresponds to the S487T mutation in SARS-CoV
has the potential to greatly enhance the binding affinity between 2019-nCoV RBD and human ACE2. Due to this, patients should be monitored closely in case there is a mutation that occurs at the 501 position. We will answer is why the binding affinity increases between 2019- nCoV RBD and human ACE2, when there is a mutation at position 501.
- What sequences will you use?
- We will use the human ACE2 sequence and 2019-nCoV RBD sequence, along with a mutated sequence, N501T.
Scientific Conclusion
this lab allowed me to explore in-depth protein structures and the different ways that it could be demonstrated in. I learned how to use software to show protein shape, interactions within a protein, and specific bonds in a protein. The skills I learned in this lab are ones that I will be using later in my individual research which we made a plan for in this lab.
Acknowledgments
- I acknowledge my partner Kam Taghizadeh discussing the questions with me.
- I used Wan et. al article as a resource for this assignment.
- I copied and modified procedures from the Week 5 assignment page.
- I copied the section "Beginning your research project" from my homework partnerKam Taghizadeh
- I acknowledge Dr. Kam D. Dahlquist, Ph.D. for giving the instructions for this assignment and helping me understand it.
- I uploaded images by using Wiki Upload page
- I found data from UniProt Knowledgebase (UniProt KB).
- I used iCn3D viewer to work on protein structures.
"Except for what is noted above, this individual journal entry was completed by me and not copied from another source."Falghane (talk) 23:05, 7 October 2020 (PDT)
References
- OpenWetWare. (2020). BIOL368/F20:Week 1. Retrieved 6 October 2020, from https://openwetware.org/wiki/BIOL368/F20:Week_1
- OpenWetWare. (2020). BIOL368/F20:Week 5. Retrieved 6 October 2020, from https://openwetware.org/wiki/BIOL368/F20:Week_5
- iCn3D: Web-based 3D Structure Viewer 3SCK. (2020). Retrieved 6 October 2020, from https://www.ncbi.nlm.nih.gov/Structure/icn3d/full.html?pdbid=%203SCK
- iCn3D: Web-based 3D Structure Viewer 2AJF. (2020). Retrieved 6 October 2020, from https://www.ncbi.nlm.nih.gov/Structure/icn3d/full.html?&mmdbid=35213&bu=1&showanno=1
- Wan, Y., Shang, J., Graham, R., Baric, R., & Li, F. (2020). Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. Journal Of Virology, 94(7). doi: 10.1128/jvi.00127-20
