AninditaVarshneya BIOL368 Week 15
Purpose
Study the dGLN3 microarray data from the Dahlquist Lab to analyze the impacts of cold shock in dGLN3 in contrast to the results found in Tai et al.
Microarray Data Analysis
- For this assignment, our group focused on dGLN3 microarray data from the Dahlquist Lab at Loyola Marymount University.
Before you begin: Turn on File Extensions
- The Windows 7 operating systems defaults to hiding file extensions. To turn them back on, do the following:
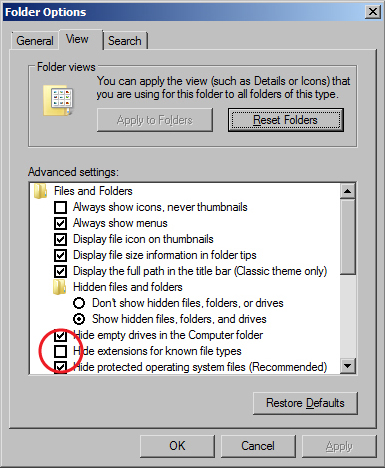
Folder Options window - Go to the Start menu and select "Control Panel".
- In the window that appears, search for "Folder Options" in the search field in the upper right hand corner.
- Click on "Folder Options" in the main window.
- When the Folder Options window appears, click on the View tab.
- Uncheck the box for "Hide extensions for known file types".
- Click the OK button.
- The computers in Seaver 120 are are set to erase all custom user settings and restore the defaults once they have been restarted, so if you have done this previously, you might have to do it again.

Experimental Design
Recall from last week's lecture, there were five timepoints in the experiment that were competitively hybridized with the t0 sample:
- t15 (cold shock) --> 4 replicates
- t30 (cold shock) --> 4 replicates
- t60 (cold shock) --> 4 replicates
- t90 (recovery) --> 4 replicates
- t120 (recovery) --> 4 replicates
- 20 replicates total.
Each timepoint has 3-5 independent replicates.
Within-strain ANOVA
The purpose of the within-stain ANOVA test is to determine if any genes had a gene expression change that was significantly different than zero at any timepoint.
- Create a new worksheet, naming it either "(STRAIN)_ANOVA" as appropriate. For example, you might call yours "wt_ANOVA" or "dHAP4_ANOVA"
- My sheet was named: "dGLN3_ANOVA"
- Copy the first three columns containing the "Master_Index", "ID", and "Standard_Name" from the "Master_Sheet" worksheet for your strain and paste it into your new worksheet. Copy the columns containing the data for your strain and paste it into your new worksheet.
- At the top of the first column to the right of your data, create five column headers of the form (STRAIN)_AvgLogFC_(TIME) where (STRAIN) is your strain designation and (TIME) is 15, 30, etc.
- dGLN3_AvgLogFC_(TIME)
- In the cell below the (STRAIN)_AvgLogFC_t15 header, type
=AVERAGE( - Then highlight all the data in row 2 associated with dGLN3 and t15, press the closing paren key (shift 0),and press the "enter" key.
- This cell now contains the average of the log fold change data from the first gene at t=15 minutes.
- Click on this cell and position your cursor at the bottom right corner. You should see your cursor change to a thin black plus sign (not a chubby white one). When it does, double click, and the formula will magically be copied to the entire column of 6188 other genes.
- Repeat steps (4) through (8) with the t30, t60, t90, and the t120 data.
- Now in the first empty column to the right of the dGLN3_AvgLogFC_t120 calculation, create the column header dGLN3_ss_HO.
- In the first cell below this header, type
=SUMSQ( - Highlight all the LogFC data in row 2 for your (STRAIN) (but not the AvgLogFC), press the closing paren key (shift 0),and press the "enter" key.
- In the next empty column to the right of dGLN3_ss_HO, create the column headers dGLN3_ss_(TIME) as in (3).
- Make a note of how many data points you have at each time point for your strain. Also, make a note of the total number of data points. Again, for most strains, this will be 20.
- There were 4 replicates per time point, and 20 replicates in total. There were 5 time points.
- In the first cell below the header dGLN3_ss_t15, type
=SUMSQ(<range of cells for logFC_t15>)-<number of data points>*<AvgLogFC_t15>^2and hit enter.- The phrase <range of cells for logFC_t15> should be replaced by the data range associated with t15.
- The phrase <number of data points> should be replaced by the number of data points for that timepoint (either 3, 4, or 5).
- The phrase <AvgLogFC_t15> should be replaced by the cell number in which you computed the AvgLogFC for t15, and the "^2" squares that value.
- Upon completion of this single computation, use the Step (7) trick to copy the formula throughout the column.
- Repeat this computation for the t30 through t120 data points. Again, be sure to get the data for each time point, type the right number of data points, and get the average from the appropriate cell for each time point, and copy the formula to the whole column for each computation.
- In the first column to the right of dGLN3_ss_t120, create the column header dGLN3_SS_full.
- In the first row below this header, type
=sum(<range of cells containing "ss" for each timepoint>)and hit enter. - In the next two columns to the right, create the headers dGLN3_Fstat and dGLN3_p-value.
- Recall the number of data points from (13): call that total n.
- In the first cell of the dGLN3_Fstat column, type
=((n-5)/5)*(<dGLN3_ss_HO>-<dGLN3_SS_full>)/<dGLN3_SS_full>and hit enter.- Don't actually type the n but instead use the number from (13). Also note that "5" is the number of timepoints and the dSWI4 strain has 4 timepoints (it is missing t15).
- Replace the phrase dGLN3_ss_HO with the cell designation.
- Replace the phrase <dGLN3_SS_full> with the cell designation.
- Copy to the whole column.
- In the first cell below the dGLN3_p-value header, type
=FDIST(ABS(<dGLN3_Fstat>),5,n-5)replacing the phrase <dGLN3_Fstat> with the cell designation and the "n" as in (13) with the number of data points total. (Again, note that the number of timepoints is actually "4" for the dSWI4 strain). Copy to the whole column.- Because we had some negative F-stat values, we needed to take the absolute values of the Fstats in order to take the p value. Without doing so, we were getting errors and were unable to calculate the p-value.
- Before we move on to the next step, we will perform a quick sanity check to see if we did all of these computations correctly.
- Click on cell A1 and click on the Data tab. Select the Filter icon (looks like a funnel). Little drop-down arrows should appear at the top of each column. This will enable us to filter the data according to criteria we set.
- Click on the drop-down arrow on your dGLN3_p-value column. Select "Number Filters". In the window that appears, set a criterion that will filter your data so that the p value has to be less than 0.05.
- Excel will now only display the rows that correspond to data meeting that filtering criterion. A number will appear in the lower left hand corner of the window giving you the number of rows that meet that criterion. We will check our results with each other to make sure that the computations were performed correctly.
- Initially, I calculated the F-stat and p-values incorrectly (with 4 time points instead of 5), so the first number I got from the sanity check was 2937. After fixing this issue, however, this total changed. The new total can be found in the data powerpoint linked here: Excel workbook containing the statistical analysis
Calculate the Bonferroni and p value Correction
- Now we will perform adjustments to the p value to correct for the multiple testing problem. Label the next two columns to the right with the same label, dGLN3_Bonferroni_p-value.
- Type the equation
=<dGLN3_p-value>*6189, Upon completion of this single computation, use the Step (10) trick to copy the formula throughout the column. - Replace any corrected p value that is greater than 1 by the number 1 by typing the following formula into the first cell below the second dGLN3_Bonferroni_p-value header:
=IF(r2>1,1,r2). Use the Step (10) trick to copy the formula throughout the column.
Calculate the Benjamini & Hochberg p value Correction
- Insert a new worksheet named "dGLN3_ANOVA_B-H".
- Copy and paste the "Master_Index", "ID", and "Standard_Name" columns from your previous worksheet into the first two columns of the new worksheet.
- For the following, use Paste special > Paste values. Copy your unadjusted p values from your ANOVA worksheet and paste it into Column D.
- Select all of columns A, B, C, and D. Sort by ascending values on Column D. Click the sort button from A to Z on the toolbar, in the window that appears, sort by column D, smallest to largest.
- Type the header "Rank" in cell E1. We will create a series of numbers in ascending order from 1 to 6189 in this column. This is the p value rank, smallest to largest. Type "1" into cell E2 and "2" into cell E3. Select both cells E2 and E3. Double-click on the plus sign on the lower right-hand corner of your selection to fill the column with a series of numbers from 1 to 6189.
- Now you can calculate the Benjamini and Hochberg p value correction. Type dGLN3_B-H_p-value in cell F1. Type the following formula in cell F2:
=(D2*6189)/E2and press enter. Copy that equation to the entire column. - Type "dGLN3_B-H_p-value" into cell G1.
- Type the following formula into cell G2:
=IF(F2>1,1,F2)and press enter. Copy that equation to the entire column. - Select columns A through G. Now sort them by your Master_Index in Column A in ascending order.
- Copy column G and use Paste special > Paste values to paste it into the next column on the right of your ANOVA sheet.
Sanity Check: Number of genes significantly changed
Before we move on to further analysis of the data, we want to perform a more extensive sanity check to make sure that we performed our data analysis correctly. We are going to find out the number of genes that are significantly changed at various p value cut-offs.
- Go to your dGLN3_ANOVA worksheet.
- Select row 1 (the row with your column headers) and select the menu item Data > Filter > Autofilter (The funnel icon on the Data tab). Little drop-down arrows should appear at the top of each column. This will enable us to filter the data according to criteria we set.
- Click on the drop-down arrow for the unadjusted p value. Set a criterion that will filter your data so that the p value has to be less than 0.05.
- How many genes have p < 0.05? and what is the percentage (out of 6189)?
- 2393 (38.7%
- How many genes have p < 0.01? and what is the percentage (out of 6189)?
- 1453 (23.5%)
- How many genes have p < 0.001? and what is the percentage (out of 6189)?
- 696 (11.2%)
- How many genes have p < 0.0001? and what is the percentage (out of 6189)?
- 320 (5.2%)
- How many genes have p < 0.05? and what is the percentage (out of 6189)?
- When we use a p value cut-off of p < 0.05, what we are saying is that you would have seen a gene expression change that deviates this far from zero by chance less than 5% of the time.
- We have just performed 6189 hypothesis tests. Another way to state what we are seeing with p < 0.05 is that we would expect to see this a gene expression change for at least one of the timepoints by chance in about 5% of our tests, or 309 times. Since we have more than 309 genes that pass this cut off, we know that some genes are significantly changed. However, we don't know which ones. To apply a more stringent criterion to our p values, we performed the Bonferroni and Benjamini and Hochberg corrections to these unadjusted p values. The Bonferroni correction is very stringent. The Benjamini-Hochberg correction is less stringent. To see this relationship, filter your data to determine the following:
- How many genes are p < 0.05 for the Bonferroni-corrected p value? and what is the percentage (out of 6189)?
- 155 (2.5%)
- How many genes are p < 0.05 for the Benjamini and Hochberg-corrected p value? and what is the percentage (out of 6189)?
- 155 (2.5%)
- How many genes are p < 0.05 for the Bonferroni-corrected p value? and what is the percentage (out of 6189)?
- In summary, the p value cut-off should not be thought of as some magical number at which data becomes "significant". Instead, it is a moveable confidence level. If we want to be very confident of our data, use a small p value cut-off. If we are OK with being less confident about a gene expression change and want to include more genes in our analysis, we can use a larger p value cut-off.
- Comparing results with known data: the expression of the gene NSR1 (ID: YGR159C)is known to be induced by cold shock. Find NSR1 in your dataset. What is its unadjusted, Bonferroni-corrected, and B-H-corrected p values? What is its average Log fold change at each of the timepoints in the experiment? Note that the average Log fold change is what we called "dGLN3_AvgLogFC_(TIME)" in step 3 of the ANOVA analysis.
- unadjusted p-value: 0.000506764
- Bonferroni-correct p-value: 3.136364678
- B-H p-value: 0.005702481
- AvgLogFC at t15: 3.506225
- AvgLogFC at t30: 4.5319
- AvgLogFC at t60: 2.7592
- AvgLogFC at t90: -1.85025
- AvgLogFC at t120: -1.867425
- Statistics data table and screenshots of the stem results
- Excel workbook containing the statistical analysis
Clustering and GO Term Enrichment with stem
- Prepare your microarray data file for loading into STEM.
- Insert a new worksheet into your Excel workbook, and name it "dGLN3_stem".
- Select all of the data from your "dGLN3_ANOVA" worksheet and Paste special > paste values into your "dGLN3_stem" worksheet.
- Your leftmost column should have the column header "Master_Index". Rename this column to "SPOT". Column B should be named "ID". Rename this column to "Gene Symbol". Delete the column named "Standard_Name".
- Filter the data on the B-H corrected p value to be > 0.05 (that's greater than in this case).
- Once the data has been filtered, select all of the rows (except for your header row) and delete the rows by right-clicking and choosing "Delete Row" from the context menu. Undo the filter. This ensures that we will cluster only the genes with a "significant" change in expression and not the noise.
- Delete all of the data columns EXCEPT for the Average Log Fold change columns for each timepoint (for example, wt_AvgLogFC_t15, etc.).
- Rename the data columns with just the time and units (for example, 15m, 30m, etc.).
- Find and replace any "!DIV/0" data in the sheet with a blank cell. These errors come up when there is a lack of data for a particular gene at a particular time point. In order for the STEM application to work correctly, we need to delete all of these values.
- Save your work. Then use Save As to save this spreadsheet as Text (Tab-delimited) (*.txt). Click OK to the warnings and close your file.
- Note that you should turn on the file extensions if you have not already done so.
- Now download and extract the STEM software. Click here to go to the STEM web site.
- Click on the download link, register, and download the
stem.zipfile to your Desktop. - Unzip the file. In Seaver 120, you can right click on the file icon and select the menu item 7-zip > Extract Here.
- This will create a folder called
stem. Inside the folder, double-click on thestem.jarto launch the STEM program.
- Click on the download link, register, and download the
- Running STEM
- In section 1 (Expression Data Info) of the the main STEM interface window, click on the Browse... button to navigate to and select your file.
- Click on the radio button No normalization/add 0.
- Check the box next to Spot IDs included in the data file.
- In section 2 (Gene Info) of the main STEM interface window, select Saccharomyces cerevisiae (SGD), from the drop-down menu for Gene Annotation Source. Select No cross references, from the Cross Reference Source drop-down menu. Select No Gene Locations from the Gene Location Source drop-down menu.
- In section 3 (Options) of the main STEM interface window, make sure that the Clustering Method says "STEM Clustering Method" and do not change the defaults for Maximum Number of Model Profiles or Maximum Unit Change in Model Profiles between Time Points.
- In section 4 (Execute) click on the yellow Execute button to run STEM.
- In section 1 (Expression Data Info) of the the main STEM interface window, click on the Browse... button to navigate to and select your file.
- Viewing and Saving STEM Results
- A new window will open called "All STEM Profiles (1)". Each box corresponds to a model expression profile. Colored profiles have a statistically significant number of genes assigned; they are arranged in order from most to least significant p value. Profiles with the same color belong to the same cluster of profiles. The number in each box is simply an ID number for the profile.
- Click on the button that says "Interface Options...". At the bottom of the Interface Options window that appears below where it says "X-axis scale should be:", click on the radio button that says "Based on real time". Then close the Interface Options window.
- Take a screenshot of this window (on a PC, simultaneously press the
AltandPrintScreenbuttons to save the view in the active window to the clipboard) and paste it into a PowerPoint presentation to save your figures.
- Click on each of the SIGNIFICANT profiles (the colored ones) to open a window showing a more detailed plot containing all of the genes in that profile.
- Take a screenshot of each of the individual profile windows and save the images in your PowerPoint presentation.
- At the bottom of each profile window, there are two yellow buttons "Profile Gene Table" and "Profile GO Table". For each of the profiles, click on the "Profile Gene Table" button to see the list of genes belonging to the profile. In the window that appears, click on the "Save Table" button and save the file to your desktop. Make your filename descriptive of the contents, e.g. "wt_profile#_genelist.txt", where you replace the number symbol with the actual profile number.
- Upload these files to OpenWetWare and link to them on your individual journal page. (Note that it will be easier to zip all the files together and upload them as one file).
- For each of the significant profiles, click on the "Profile GO Table" to see the list of Gene Ontology terms belonging to the profile. In the window that appears, click on the "Save Table" button and save the file to your desktop. Make your filename descriptive of the contents, e.g. "wt_profile#_GOlist.txt", where you use "wt", "dGLN3", etc. to indicate the dataset and where you replace the number symbol with the actual profile number. At this point you have saved all of the primary data from the STEM software and it's time to interpret the results!
- Upload these files to OpenWetWare and link to them on your individual journal page. (Note that it will be easier to zip all the files together and upload them as one file).
- A new window will open called "All STEM Profiles (1)". Each box corresponds to a model expression profile. Colored profiles have a statistically significant number of genes assigned; they are arranged in order from most to least significant p value. Profiles with the same color belong to the same cluster of profiles. The number in each box is simply an ID number for the profile.
- Analyzing and Interpreting STEM Results
- Select one of the profiles you saved in the previous step for further intepretation of the data. I suggest that you choose one that has a pattern of up- or down-regulated genes at the cold shock timepoints. Each member of your group should choose a different profile. Answer the following:
- Why did you select this profile? In other words, why was it interesting to you?
- I selected this profile because it had the most significant p-value and because it shows quite a bit of down regulation between t30-t90. I am interested to see what is being down regulated, and what that means in the context of the cell.
- How many genes belong to this profile?
- 446 genes were assigned to this profile
- How many genes were expected to belong to this profile?
- 35.88 genes were expected to be assigned to this profile.'
- What is the p value for the enrichment of genes in this profile? Bear in mind that we just finished computing p values to determine whether each individual gene had a significant change in gene expression at each time point. This p value determines whether the number of genes that show this particular expression profile across the time points is significantly more than expected.
- The p-value for the enrichment of genes in this profile is 0.00 (significant).
- I chose profile 45, Isai chose profile 7, Will chose profile 2, and Shivum chose profile 48.
- Open the GO list file you saved for this profile in Excel. This list shows all of the Gene Ontology terms that are associated with genes that fit this profile. Select the third row and then choose from the menu Data > Filter > Autofilter. Filter on the "p-value" column to show only GO terms that have a p value of < 0.05. How many GO terms are associated with this profile at p < 0.05? The GO list also has a column called "Corrected p-value". This correction is needed because the software has performed thousands of significance tests. Filter on the "Corrected p-value" column to show only GO terms that have a corrected p value of < 0.05. How many GO terms are associated with this profile with a corrected p value < 0.05?
- GO list for Profile 45 as Excel Spreadsheet
- 250 GO terms are associated with this profile at p-value < 0.05.
- 79 GO terms are associated with this profile at a corrected p-value <0.05.
- Select 6 Gene Ontology terms from your filtered list (either p < 0.05 or corrected p < 0.05).
- The terms I am considered include:
- ribosome biogenesis, GO:0042254
- nucleolus, GO:0005730
- ncRNA processing/ metabiolic process/ RNA metabolic process GO:0034660
- nucleic acid metabolism GO: 0090304
- organic cyclic compound metabolic process/ cellular aromatic compound metabolic process GO: 1901360
- maturation of 5.8S rRNA from tricistronic rRNA transcript (ssu-rRNA, 5.8s rRNA, LSU-rRNA) GO: 0000466
- Each member of the group will be reporting on his or her own cluster in your presentation next week. You should take care to choose terms that are the most significant, but that are also not too redundant. For example, "RNA metabolism" and "RNA biosynthesis" are redundant with each other because they mean almost the same thing.
- Note whether the same GO terms are showing up in multiple clusters.
- Look up the definitions for each of the terms at http://geneontology.org. In your final presentation, you will discuss the biological interpretation of these GO terms. In other words, why does the cell react to cold shock by changing the expression of genes associated with these GO terms? Also, what does this have to do with the transcription factor being deleted (for the Δgln3 and Δswi4 groups)?
- ribosome biogenesis: a cellular process that results in biosynthesis of constituent macro-molecules, assembly, and arrangement of constituent parts of ribosome subunits.
- nucleolus: small dense body rich in RNA and proteins, not bound by limiting membrane, prime function is transcription of nuclear DNA to 45S ribosomal precursor RNA and processing RNA into 5.8S, 18S, and 28S components.
- ncRNA metabolic process: chemical reactions and pathways involving non-coding RNA transcripts.
- nucleic acid metabolic process: any cellular metabolic process involving nucleic acids.
- organic cyclic compound metabolic process: chemical reactions and pathways involving organic cyclic compounds.
- maturation of 5.8 rRNA from tricistronic rRNA transcript: any process involved in maturation of an rRNA molecule originally produced as tricistronic rRNA transcript that contained the small subunit rRNA, the 5.8 rRNA and the large subunit rRNA.
- These processes are related to the deletion of the gln3 transcription factor because this means that gln3 is not involved in these processes. More substantial findings can be determined if we analyze the difference between our data and the data collected by the group analyzing the wild-type data.
- To easily look up the definitions, go to http://geneontology.org.
- Copy and paste the GO ID (e.g. GO:0044848) into the search field at center top of the page called "Search GO Data".
- In the results page, click on the button that says "Link to detailed information about <term>, in this case "biological phase"".
- The definition will be on the next results page, e.g. here.
- The terms I am considered include:
- Why did you select this profile? In other words, why was it interesting to you?
- Select one of the profiles you saved in the previous step for further intepretation of the data. I suggest that you choose one that has a pattern of up- or down-regulated genes at the cold shock timepoints. Each member of your group should choose a different profile. Answer the following:
Data and Files
- Excel workbook containing the statistical analysis
- The input file for stem
- Zipped _genelist.txt files from stem
- Zipped _GOlist.txt files from stem
- GO list for Profile 45 as Excel Spreadsheet
- Statistics data table and screenshots of the stem results.
- Final Presentation
Conclusion
This lab allowed us to analyze the impacts of cold shock on a strain of S. cerevisiae with transcription factor gln3 deleted. We analyzed these results using the STEM software. I analyzed profile 45 for any significant processes affected by cold shock. Ribosomal biogenesis was up-regulated during the cold shock time points and down-regulated during recovery time points. This is most likely because additional proteins need to be produced during cold shock stress to help the species maintain homeostasis. These results were reflected in Tai et al. as well as Infanzon et al. Other key findings were that mitosis and nitrogen compound catabolism were both regulated during cold shock conditions as well.
Acknowledgements
My group, Will Fuchs, Isai Lopez, and Shivum Desai, worked together to complete all aspects of this assignment in class on 12/6, and outside of class on 12/11 and 12/12 for several hours. Thank you to Dr. Kam D. Dahlquist for helping me complete this assignment. The procedures from this assignment were copied from the Week 15 assignment page. While I worked with the people noted above, this individual journal entry was completed by me and not copied from another source.
References
- Cai, L., & Tu, B. P. (2012). Driving the cell cycle through metabolism. Annual review of cell and developmental biology, 28, 59-87. doi: 10.1146/annurev-cellbio-092910-154010
- Ermolenko, D. N., and G. I. Makhatadze. "Bacterial cold-shock proteins." Cellular and Molecular Life Sciences CMLS 59.11 (2002): 1902-1913. doi: 10.1128/jb.171.5.2337-2346.1989
- Infanzon, B. A., Buckmelter, K. M., Liu, E. M., Sakhon, O. S., Citti, W. T., & Dahlquist, K. D. (2010). Saccharomyces cerevisiae responds to cold shock by inducing the transcription of ribosome biogenesis genes. The FASEB Journal, 24(1 Supplement), 833-13.
- Tai, S. L., Daran-Lapujade, P., Walsh, M. C., Pronk, J. T., & Daran, J. M. (2007). Acclimation of Saccharomyces cerevisiae to low temperature: a chemostat-based transcriptome analysis. Molecular Biology of the Cell, 18(12), 5100 5112. doi: 10.1091/mbc.E07-02-0131
- Zhu, XP., You, F., Zhang, PJ. et al. Mar Biotechnol (2006) 8: 312. doi:10.1007/s10126-006-5128-3
- Week 15 Assignment
Other Links
User Page: Anindita Varshneya
Bioinfomatics Lab: Fall 2016
Class Page: BIOL 368-01: Bioinfomatics Laboratory, Fall 2016
| Weekly Assignments | Individual Journal Assignments | Shared Journal Assignments |
|---|---|---|
|
|
|
SURP 2015
Links: Electronic Lab Notebook
