BIOL368/F14:Chloe Jones Week 8
Defining Your HIV Structure Research Project
Project Partners: Isabel Gonzaga and Nicole Anguiano Chose to analyze Isabel's question into further detail, information below is taken from Isabel Gonzaga Week 8
Question
How does HIV status (diagnosed, progressing or non-trending) affect the structure of the V3 protein region?
Hypothesis
We hypothesize that diagnosed groups will express greater variability in the V3 region in their protein structure, in comparison to the non-trending groups. Initial comparisons show that diagnosed groups and progressing groups expressed greater genetic variability than non-trending groups. These changes may affect the third variable region, affecting the host's ability to adapt to the changes and generate sufficient immune response.
Subject Data
According to the BEDROCK HIV Sequence Data Table, I was able to determine which of the subjects used within my study actually developed aids. All 3 AIDS diagnosed were confirmed with the disease by their final visit. In the AIDS progressing groups, subjects developed AIDS within 1 year after their final visit. The Non-Trending groups all maintained high CD4 T Cell Counts above the threshold, even after the study was conducted. Sequences were for each visit and subject were chosen using a Random Integer Generator, to eliminate selection bias.
The following sequences was taken from the BEDROCK HIV Problem Space Database, from the Markham et al. (1998) study.
Table 1: Sequences analyzed
| Group | Subject | Visit | Sequences |
|---|---|---|---|
| AIDS Diagnosed | 3 10 15 |
1 6 1 6 1 4 |
1, 2, 4 3, 4, 5 3, 6, 7 2, 4, 8 2, 3, 4 5, 8, 10 |
| AIDS Progressing | 7 8 14 |
1 5 1 7 1 9 |
2, 3, 9 2, 8, 9 1, 4, 5 1, 6, 7 2, 3, 4 9, 10, 11 |
| No Trend | 5 6 13 |
1 5 1 9 1 5 |
1, 3, 8 4, 5, 2 1, 2, 3 6, 7, 9 1, 3, 4 3, 5, 4 |
Working with Protein Sequences In-class Activity
Chapter 4: Reading a SWISS-PROT entry
Source: Bioinformatics for Dummies pp.110-123
- Using the database UniProt KB which has two subsections: Swis-Prot and TrEMBL.
- If you search on the keywords "HIV" and "gp120", how many results do you get?
- 180,227 results.
General Information about the entry
- Entry name: 9HIV1
- Primary Accession Number :Q75760
- Secondary Accession Numbers :N/A
- Intergrated into Swiss-Prot on :November 1, 1996
- Sequence was last modified on : November 1, 1996
- Annotations were last modified on : October 1, 2014
Name and origin of the protein
- Protein name:Envelope glycoprotein gp160
- Synonyms:N/A
- Gene name:Env
- From:Homo sapiens (Human) [TaxID: 9606] TaxID:9606
- Taxonomy: Viruses › Retro-transcribing viruses › Retroviridae › Orthoretrovirinae › Lentivirus › Primate lentivirus group
References
- Number of references:9
- The cross references:172
- EMBL: Coding Sequence U63632.1
- PDB Crystal Structures: Entry 2b4c
- Modbase: theoretically calculated model database
- DIP (Database of Interacting Proteins): DIP-59960N
- IntAct: Q75760. 3 interactions observed
- Interpro
- PFam
- EMBL: Coding Sequence U63632.1
Keywords
- Apoptosis, fushion of virus membrane with host membrane, host-virus interaction, viral attachment to host cell, viral immunoevasion, Viral penetration into host cytoplasm, virus entry into host cell
The Sequence
- Q75760 : FASTA format

Chapter 5: ORFing your DNA sequence
Source: Bioinformatics for Dummies pp.146-147
- Fasta format of Envelope glycoprotein gp160 obtained from UniProt KB was placed in the in the imput box of NCBI Open Reading Frame Finder . Six parallel horizontal bars were present with integers correlating to the reading frame.
- Putting in DNA sequences: Subject 1 visit 1, clone 1:
- The protein with the open reading frame was in the ORF Finder database was projected to be #2, or #3. Looking at the exPASy database 5’3” Frame 3 had an open reading frame with no stop codons interrupting /truncating the lengh of the DNA.
Chapter 6:Working with a single protein sequence
Source: Bioinformatics for Dummies pp.159-195
Predicting the main physico-chemical properties of a protein
- Used the program ExPasy ProtParam for computation of physcial and chemical parameters of a given protein
- Imput the Swiss-Prot/TrEMBL accession number Q75760. Then click compute parameters button >Submit
- Corresponds to the HIV gp120 sequence that was used for the crystal structure for the Huang et al. (2005) paper.
- Can also enter raw sequence
- Save the file
Interpreting ProtParam results
- Used ExPasy ProtParam for data.
- Molecular Weight: 96160.4 Daltons
- Extinction Coefficients:
- assuming all pairs of Cys residues form cystines= 184145M1cm1
- assuming all Cys residues are reduced= 182770 M1cm1
- ~Tell you how much light (visible or invisible) your protein absorbs at a certain wavelength.
- Instability
- instability index (II) is computed to be 37.91
- classifies the protein as stable
- Half-Life
- The estimated half-life is:
- 30 hours (mammalian reticulocytes, in vitro)
- >20 hours (yeast, in vivo)
- >10 hours (Escherichia coli, in vivo)
- The estimated half-life is:
Digesting a protein in a computer
- Used ExPasy Peptide Cutter
- Imput accession number: Q75760
- Program cuts protein at specific site
Doing primary structure analysis Looking for transmembrane segments
- Used http://web.expasy.org/cgi-bin/protscale/protscale.pl ExPasy Protscale]
- Imput accession number: Q75760
- The amino acid scale Hphob. / Kyte & Doolittle was preselected
- Changed the Window size to 19 because best window value for viewing trans membrane regions
- Covert the image to GIF format
- Save file
- Interpreting ProtScale results
- To confirm use Hphob. / Eisenberg et al.scale, set threshold to 1.6
- Piece of paper to over results
- lower paper to strongest peaks visible
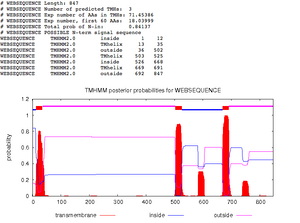
Figure 4. Identifying transmembrane domains using the TMHMM database. Transmembrane proteins are denoted in red. - Keep lowering threshold as long as you keep seeing sharp peeks
- 4 Sharp Peaks, 4 Transmembrane domains
- Running TMHMM
- UseTMHMM Q75760 sequence was inputted in FASTA format>Submit
- 5 transmembrane domains identified Figure. 3
- Looking for coiled regions
- Used COILS server at EMBnet, input accession number Q75760
- Changed input sequence format to SwissProtID or AC
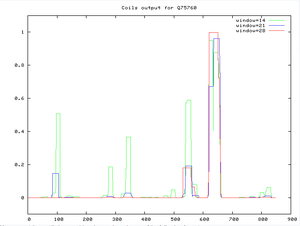
Figure 5. Coils output for Q75760, using the coils server at EMBnet. Look at regions between 600-700.


Predicting post-translational modifications in your protein
- Looking for PROSITE patterns
- Used ScanProsite, input accession number Q75760
- Uncheck excluded motifs with high probability of occurrence, check the excluded profiles from scan box >start the scan
- Interpreting ScanProsite results

- Slide mouse over color rectangles to see name displayed, click to receive more information
- the list of segments contain patterns within protein; numbers indicate position and capital letters are specified and lowercase letters are unspecified by patterns
- ~be careful with short patterns
- Weak signals add up to give strong signals –two related patterns at close distance, significant
- Eliminating weak patterns-multiple sequence alignment
Finding Known Domains in Your Protein
- Finding Domains with InterProScan
- InterProScan, imput fasta format of Q75760 sequence>submit

- Finding domains with the CD server
- Used NCBI CD Server, input accession number Q75760,
- Deselect the apply low complexity filter check box, change threshold set to 1 >Submit Query'
- Interpreting and Understanding CD results
- Red domain are from SMART
- Ragged indicates partial matches
- lower e-value correlates to a better score

- Finding domains with Motif Scan
- Unfortunately, the the Motif-Scan provider was having complications with generating the information upon putting in the FASTA format.
Chapter 11: Predicting the secondary structure of a protein sequence and additional structural features
Source: Bioinformatics for Dummies pp.330-336
From the Primary to Secondary Structures
- Predicting the secondary structure of a protein sequence
- Used PsiPred, input protein sequence S1V1-1 via FASTA format, and gave the sequence a short identifier (s1v1-1).
After the job was submitted it ____minutes to generate the information

- Predicting additional structural features
- Used Predict Protein. In order to use program had to make an account. Input amino acid for S1V1-1.

Comparison of crystal structure of gp120 From the NCBI website the Hunag et al. (2005) paper was downloaded via CN3D file.To make it more visible I used a secondary shortcut to make the helices (green) and beta sheets (yellow) so they could be better differentiated and analyzed. The structure of the protein form NCBI website favors a composition favors a composition where there are more beta sheets present with random coil in comparison to the prediction from PsiPred that had them being in equal abundance.

Electronic Lab Notebook
- Chloe Jones Week 2
- Chloe Jones Week 3
- Chloe Jones Week 4
- Chloe Jones Week 5
- Chloe Jones Week 6
- Chloe Jones Week 7
- Chloe Jones Week 8
- Chloe Jones Week 9
- Chloe Jones Week 10
- Chloe Jones Week 11
- Chloe Jones Week 12
- Chloe Jones Week 13
- Chloe Jones Week 15
Weekly Assignments
- Week 1 Assignment
- Week 2 Assignment
- Week 3 Assignment
- Week 4 Assignment
- Week 5 Assignment
- Week 6 Assignment
- Week 7 Assignment
- Week 8 Assignment
- Week 9 Assignment
- Week 10 Assignment
- Week 11 Assignment
- Week 12 Assignment
- Week 13 Assignment
- Week 15 Assignment
Class Journals
- Class Journal Week 1
- Class Journal Week 2
- Class Journal Week 3
- Class Journal Week 4
- Class Journal Week 5
- Class Journal Week 6
- Class Journal Week 7
- Class Journal Week 8
- Class Journal Week 9
- Class Journal Week 10
- Class Journal Week 11
- Class Journal Week 12
- Class Journal Week 13
- Class Journal Week 15
Chloe Jones 03:46, 15 October 2014 (EDT)Chloe Jones
