Alex A. Cardenas Week 3
From OpenWetWare
Jump to navigationJump to search
Activity 1
Methods
- Went to pubmed to find the Markham paper
- Clicked on nucleotide under the links to find the sequences
- Retrieved the sequence file S4V2-4 and viewed it in the FASTA format
- Which was then copy and pasted into Biology workbench.
- Copy and pasted sequences into a txt. file saved it as 'sequence.txt'
- Uploaded all of the files and then ran them all using ClustalW
Results
- After running the program and sequences compared, the scores were:
Sequences (1:2) Aligned. Score: 86 Sequences (1:3) Aligned. Score: 85 Sequences (1:4) Aligned. Score: 97 Sequences (1:5) Aligned. Score: 97 Sequences (2:3) Aligned. Score: 97 Sequences (2:4) Aligned. Score: 84 Sequences (2:5) Aligned. Score: 84 Sequences (3:4) Aligned. Score: 83 Sequences (3:5) Aligned. Score: 84 Sequences (4:5) Aligned. Score: 96
- These scores indicate that when compared to eachother, AF016768.2, AF0106762.2, and AF016818.2 are much more similiar to each other than they are to AF084141.1 and AF089142.1; as seen in the tree diagram below.
- The unrooted tree was
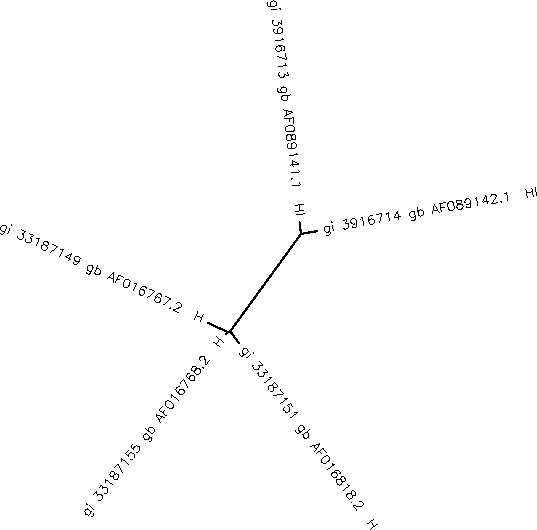

Questions
- What was the accession number of the sequence you chose?
- AF016818
- Which subject of the study was that HIV sequence from? Which section of the record contains information about who the HIV was collected from?
- Subject 2 visit 4
- Envolope glycoprotein region V3 (env) gene
Activity 2
Part 1 Methods
- Started by uploading the "visit_1_S1_S9.txt" and "visit_1_S10_S15.txt" into workbench
- Picked 12 sequences out of the 97.
S1-1 S1-4 S1-7 S2-1 S2-4 S2-6 S9-1 S9-3 S9-5 S14-2 S14-5 S14-6
- The alignment scores, the higher the score the higher the pairing/similarities, are as follows.
Aligning... Sequences (1:2) Aligned. Score: 99 Sequences (1:3) Aligned. Score: 99 Sequences (1:4) Aligned. Score: 90 Sequences (1:5) Aligned. Score: 90 Sequences (1:6) Aligned. Score: 91 Sequences (1:7) Aligned. Score: 92 Sequences (1:8) Aligned. Score: 92 Sequences (1:9) Aligned. Score: 93 Sequences (1:10) Aligned. Score: 93 Sequences (1:11) Aligned. Score: 91 Sequences (1:12) Aligned. Score: 92 Sequences (2:3) Aligned. Score: 99 Sequences (2:4) Aligned. Score: 90 Sequences (2:5) Aligned. Score: 90 Sequences (2:6) Aligned. Score: 91 Sequences (2:7) Aligned. Score: 92 Sequences (2:8) Aligned. Score: 92 Sequences (2:9) Aligned. Score: 93 Sequences (2:10) Aligned. Score: 93 Sequences (2:11) Aligned. Score: 91 Sequences (2:12) Aligned. Score: 92 Sequences (3:4) Aligned. Score: 90 Sequences (3:5) Aligned. Score: 90 Sequences (3:6) Aligned. Score: 91 Sequences (3:7) Aligned. Score: 92 Sequences (3:8) Aligned. Score: 92 Sequences (3:9) Aligned. Score: 93 Sequences (3:10) Aligned. Score: 93 Sequences (3:11) Aligned. Score: 91 Sequences (3:12) Aligned. Score: 92 Sequences (4:5) Aligned. Score: 99 Sequences (4:6) Aligned. Score: 99 Sequences (4:7) Aligned. Score: 89 Sequences (4:8) Aligned. Score: 89 Sequences (4:9) Aligned. Score: 90 Sequences (4:10) Aligned. Score: 90 Sequences (4:11) Aligned. Score: 89 Sequences (4:12) Aligned. Score: 89 Sequences (5:6) Aligned. Score: 99 Sequences (5:7) Aligned. Score: 89 Sequences (5:8) Aligned. Score: 89 Sequences (5:9) Aligned. Score: 90 Sequences (5:10) Aligned. Score: 90 Sequences (5:11) Aligned. Score: 89 Sequences (5:12) Aligned. Score: 89 Sequences (6:7) Aligned. Score: 90 Sequences (6:8) Aligned. Score: 90 Sequences (6:9) Aligned. Score: 90 Sequences (6:10) Aligned. Score: 90 Sequences (6:11) Aligned. Score: 90 Sequences (6:12) Aligned. Score: 90 Sequences (7:8) Aligned. Score: 99 Sequences (7:9) Aligned. Score: 99 Sequences (7:10) Aligned. Score: 98 Sequences (7:11) Aligned. Score: 97 Sequences (7:12) Aligned. Score: 98 Sequences (8:9) Aligned. Score: 99 Sequences (8:10) Aligned. Score: 98 Sequences (8:11) Aligned. Score: 97 Sequences (8:12) Aligned. Score: 98 Sequences (9:10) Aligned. Score: 99 Sequences (9:11) Aligned. Score: 97 Sequences (9:12) Aligned. Score: 98 Sequences (10:11) Aligned. Score: 97 Sequences (10:12) Aligned. Score: 98 Sequences (11:12) Aligned. Score: 98*Used the ClustalW tool to build alignments and distance-based unrooted trees.
- For the most part the similarities stayed mainly within all of the 4 subjects individually. However, the scores and tree diagrams showed that subject 1 and subject 2 portrayed similarities within their base pairs.
Questions
- Do the clones form each subject cluster together?
- Yes, as seen in the tree diagram below, each subjects clones cluster together.

- Do some subjects' clones show more diversity than others?
- Yes, although all of the subjects clones seem to be very closely related to other clones within the subject, the clones from subjects 1, 2, and 9 seem to be very closely related. In fact 2 of the clones have other clones branching off from it, implying that their base pairing is very similar.
- Do some of the subjects cluster together?
- Yes, the only subjects that clustered together were subjects 1 and 2.
- Write a brief description of your tree and how you interpret the clustering pattern with respect to the similarities and potential evolutionary relationships between subjects HIV sequences.
- Only four out of the fifteen subjects were tested in this run. From seeing the tree and the clustering pattern, it is likely to conclude that maybe subjects of earlier numbers were more closely related than those of higher subjects.
Part 2 Methods
Quantifying diversity within and between subjects
- For this section, all clones from 3 subjects were individually ran and aligned.
- Calculated S by counting the number of positions where at one nucleotide was different across the clones.
- Table three shows an analysis for all three subjects.
Table 3 Subject | # of Clones | S | Theta |Min Difference | Max Difference 10 7 7 2.86 1.14 3.135 15 12 28 7.2 5.076 23.97 11 7 8 3.08 0.003 0.024
- Theta is an estimate of the average pairwise genetic difference and was calculated using the formula: Theta=S / Summation (n-1) (1/i)
Links
- Alex A. Cardenas
- Week 3 Assignment
- Alex A. Cardenas Week 2
- Alex A. Cardenas Week 3
- Alex A. Cardenas Week 4
- Alex A. Cardenas Week 5
- Alex A. Cardenas Week 6
- Alex A. Cardenas Week 7
- Alex A. Cardenas Week 8
- Alex A. Cardenas Week 9
- Alex A. Cardenas Week 10
- Alex A. Cardenas Week 11
- Alex A. Cardenas Week 12
- Alex A. Cardenas Week 13
- Alex A. Cardenas Week 14
- Class Journal Week 1
- Class Journal Week 2
- Class Journal Week 3
- Class Journal Week 5
- Class Journal Week 6
- Class Journal Week 7
- Class Journal Week 8
- Class Journal Week 9
- Class Journal Week 10
- Class Journal Week 11
- Class Journal Week 12
- Class Journal Week 13
- Class Journal Week 14
