2020(S12) Lecture:week 2

Week 2 Tuesday
E. chromi

Now think about building a DNA program that runs a cell. We saw last week how a yeast gene could be moved to bacteria, allowing those cells to smell like bananas under certain growth conditions. From this one experiment you might be inclined to think that DNA parts will run reliably, independent their cellular context. Today you will explicitly test that notion.
Genetic Programs
You will compare the behavior of two genetic programs:
These genetic programs were designed, constructed, and tested by the 2009 University of Cambridge iGEM team.
Cellular Chassis
In small groups, you will put these programs into two kinds of E. coli
- Strain 4-1, a K-12 strain of E. coli
- Strain 4-2, a B-type strain.
If you are curious about the ancestry of these two "breeds," there is an interesting article linked here. The short version of the story is that the K-12 strain was isolated from a stool sample of a dipthertia patient in Palo Alto, CA and domesticated for the lab by Tatum and Lederberg around 1922. By contrast, a B-type strain was used in the early 1900s by the Phage Group at the Institute Pasteur. Both types of E. coli are still widely used in research labs today.
Procedure
- Begin by reviewing BioPrimer 7. If you'd prefer you can download it here.Is it clear what the differences are between the 2 strains of bacteria we'll be studying? What about the genetic programs?
- Next we'll watch a short animation about the technique of DNA transformation. Is it clear what the steps are and why they are performed?
- Finally, you'll work in small groups to transform the purple or the green color generators into Strains 4-1 or 4-2 as described here. A pdf of your protocol is here.
- When you are done performing these manipulations, please wash your hands.
- The petri dishes will be incubated at 37° overnight and you will examine the results of your work tomorrow.
- Before you leave today, we'll consider these questions:
- Did you make any mistakes that might affect the outcome of this experiment?
- How confident are you in the results you'll see tomorrow?
- Are you expecting colonies on all the plates? Are you expecting the same numbers on all plates?
- Are you expecting the colonies to all look the same?
- If there are differences tomorrow, how will you explain them?
- If there are differences tomorrow, what could you do to test your explanations?
|
You've taken some seemingly simple steps today and done something pretty awesome, namely intentionally imbued a bacterial host with properties you've chosen. Tomorrow, if all has gone well, you'll see colorful, antibiotic-resistant bacteria growing on the petri dishes. This transformation technology has been a routine lab procedure for a generation or so. |
Week 2 Studio
E. chromi, Day 2
You'll remember that yesterday we transformed the purple or the green color generators into Strains 4-1 or 4-2. Record the following data:
![]()
Consider again these questions from yesterday:
- Did any mistakes you made seem to affect the outcome of this experiment?
- How accurately did you predict the outcome you see?
- Do you see colonies on the plates you expected? Do you see the same numbers of colonies on all plates?
- Do all the colonies look the same?
- If you see differences, how can you explain them?
- If you see differences, what could you test your explanations?
Before we leave this exercise about chassis effects, upload your data to the BioBuilder website, login = "natbioethics"
Introducing Synthia!

Next, let's consider in detail the primary journal article that describes this technical feat and the reactions to it. The review slides for today are here. Our conversation will cover:
- the technical accomplishment itself (May 2010)
- the reaction to the announcement, including Barack Obama's call for a BioEthics Panel to review the field (May 2010)
- the work of the BioEthics Panel, including the procedures for gathering information and the recommendations that the panel ultimately offered (December 2010)
- reactions of government, scientists and watchdog groups to the recommendations
|
These are current events that will affect the progress of the field of synthetic biology. The events model how a civil society can address contentious issues. We'll look back at historical examples of such issues when we consider Asilomar, recombinant DNA technologies and the Cambridge City Council hearings. Finally, this lesson should inform your thinking and help you plan your work in 20.020 and your life in 2020 and beyond. |
Your Turn!
Last week you chose some areas for biotechnology that seemed important/interesting to you. These areas were:
- Food or Energy
- Environment
- Health or Medicine
- Manufacturing
- New Application Area
- Foundational Advances
- Information Processing
You will be grouped now according to areas of common interests. Once in your groups you should do the following:
First, introduce yourselves if you don't already know eachother.
Second, appoint a note-taker in the group. The notetaker can use the pads of papers at your tables, the white boards, or the back of the sheet listing your temporary groups
Next, talk about any topics in your area that you find especially interesting. These could be motivated by an article you've read, a personal experience, a research project you know about. Be sure everyone speaks, even if it's to say that no ideas really jump out just yet. You will need at least three ideas on the list, though, before leaving today. Ideally there should be 5 or 10 ideas.
Finally, list your ideas on the back of the sheet with your temporary groups. For example, last year the environmental groups listed:
- VACCINE (better storage, no refrigeration, esp in developing countries)
- ENVIRONMENTAL SENSORS
- SUSTAINABLE CROPS
After a while (and before 4PM) the 20.385 students will get pulled out of your groups to discuss a journal article. As 20.20ers you are welcome to:
- hand in your sheet and leave for the day, or
- continue to brainstorm ideas to add to the list, then go to option #1, or
- listen to the following podcast about the ethical implications of synthetic biology projects. The podcast file itself can be accessed here. It is about 1/2 an hour long. As you listen, take can think about how ethics might impact the projects you collected. If you'd like, take a few minutes to rank the projects areas you've considered:
- List one: topics of greatest interest to you or your group
- List two: topics that raise the greatest ethical concerns
Week 2 Thursday
U-do-it: DNA construction, recombinant DNA and PCR

Warm up: Restriction enzymes to clone a paper gene
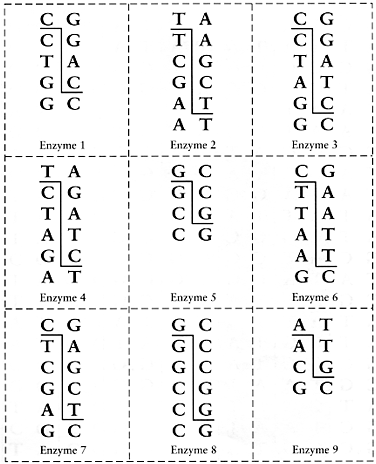
This exercise is adapted from access excellence. You will need a yellow, blue and white page ahowing DNA sequences and enzyme recognition sites.
- Begin by cutting the blue page into strips and taping the strips together according to the numbers at the bottom of each strip. In the end you will have a line (not a circle) of paper, with a gray shaded section within it. This gray section is the gene you'd like to clone from some genomic DNA.
- Next scan this blue strip of paper for the enzyme restriction sites. In the interest of time today, please only identify on the blue strip of sequence where you can find the DNA recognition sequence for Enzyme 3, Enzyme 6 and Enzyme 9. When you find them, you can mark on the paper where the two strands of DNA would be divided by the enzyme, and also mark the enzyme number.
- Next cut the yellow page into the indicated strips, tape the strips together in any order, and finally tape the ends together to make a loop. This is the plasmid vector where you will "clone" your gene from the blue paper.
- On the yellow circle of sequence, find the recognition sites for Enzymes 3, 6 and 9.
- Assuming you found some enzyme combination that flanks the blue paper gene and also exists in the yellow paper circle, cut along the lines you've drawn and combine the DNA elements.
- Note 1: the yellow circle must include the small gray box since that symbolizes elements needed for the plasmid to survive, e.g. the replication origin and the antibiotic marker.
- Note 2: the blue sequence must contain all of the gene you are trying to clone. If you choose an enzyme that cuts inside of the gene then you will be losing information that is needed for its expression. Choose a different pair of enzymes if you can.
- Note 3: were you able to "orient" your blue gene in the yellow plasmid or could it be cloned in either orientation?
- Viola! You've cloned your gene...as least on paper.
The main event: cloning pPRL or pGRN
Let's now turn to a piece of DNA that you've been working with already, namely the genes that are cloned on the pPRL and the pGRN plasmids. To re-clone these, we'll need more than paper, scissors and tape. We'll need to know some real enzymes and we'll need some actual DNA. Enzymes can be found most easily at the website of a local company, New England Biolabs, NEB.

- Start by going to the NEB homepage and typing in the DNA sequence for Enzyme 3, namely GGATCC. What is the identity of this enzyme? Repeat this for paper enzyme 6.
- Next, we'll find out what restriction sites are present at the ends of the pPRL and the pGRN gene clusters. To figure this out, let's use the Registry of Standard Biological Parts. In the homepage search bar type in the part number for either pPRL = BBa_K274002, or pGRN = BBa_K274004.
- Once you have been taken to the part page for those DNA functions, scroll down to the "sequence and features" diagram (shown above) and click the link on the right that says "Get selected sequence."
- WOW! That's a lot of Gs, As, Ts, and Cs...definitely don't want to be looking for restriction sites the way we did for the paper versions of the gene. Luckily you can copy and paste the sequence into a search tool provided by NEB called NEB cutter.
- Having done that for your pPRL or pGRN sequence, can you find an enzyme that flanks the genes?
- OK...now you'll need someplace to put the gene. Go back to the NEB cutter tool, clear any sequences that might be in the search box and then use the drop-down menu from the right tab to find a plasmid backbone. In "real life" you'd have some reason to choose one kind of destination plasmid over another, but for today you can randomly choose one to clone your purple expression cassette into.
- Once you press "submit" a drawing of a circular plasmid will show up on your screen and all the single cutter restriction sites will be labeled.
- Here's the $64,000 question: are the restriction sites you chose for the pPRL or pGRN genes on that plasmid. If so, then YAY! that was easy! But if not, what do you do? Maybe choose a new plasmid and hope for a better outcome? Or maybe we can figure out a way to put the needed restriction sites onto the pPRL or pGRN genes. Look next to PCR
The Polymerase Chain Reaction
After a quick review for how this technique works and how it can be used to introduce special restriction sites, you'll have a chance to try it yourself.
Option 1
- Use a toothpick to mix one colony of either pPRL or pGRN from your transformation plate into 100 ul of water.
- Move 5 ul of that mixture to a tube that has 100 ul of PCR mix in it.
Option 2
- Add 5 ul of dilute DNA to a tube with 100 ul of PCR mix in it.
Next steps
The reactions will be placed in a thermal cycler to heat and cool and hopefully make more of the DNA you need with the needed restriction sites on the ends. We'll come back to look at this DNA in a few weeks.
|
Part 1: Before Friday, please add some thoughts to the class blog. This will be the first of 6 entries you'll make this term. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}