Lab Notebook - Susan
October
10-10-09
- Results of dot blot: 3rd set of experiments

Unnormalized. Displayers+scFv on the left and Displayers only on the right.
usedT0.5ul of cell lysate. block 45min. 3X washes. antibody incubated 1hr. 1:500 ab:TBST.
There are no signals on the no pass. There is some background and also the negative controls also have a bit of signal. However, some of the constructs had definite signals that were a lot darker than the other signals. namely, yuaQ and upaG_short. of the three dot blots done, 3 constructs were consistent-ish hits: AIDA, espP, and cpg_l2. YuaQ, azo, and ecpx were hits on 2nd and 3rd blot (they were omitted on the first blot). 3 constructs were hits on the first and 3rd blot: hia, hag, and upaG_short.
10-6-09
- Dot blot #2

these results show hits on: ehab, vta, opr, ecpx, yua, azo, cpg, espp, and AIDA. Also note that the negative controls did not have significant signal.
10-5-09
protocol for the dot blot 10-5-09
- analyze results from first dot blot
OD600 measurements for 10-2-09 dot blot experiment

some possible hits: hag, hia, upaG_short,CPG_L2, espP, possibly AIDA. there is also one dot on what should be 1363. Not sure if that is significant.
there is no significant difference between ODs of the negative controls and the Needle scFv constructs that showed on the dot blot.
10-1-09
- ran a test BCA assay with inoculated cells. Initial OD was taken. Cells were spun down and the LB flicked out. Resuspended in 2% SDS and incubated at 50C for 30minutes. put in various amounts of cell lysate: 20ul to 200ul of reagents (10x dil), and 2ul to 198ul of reagents (100X dil). Also ran standard with BSA diluted to 1ml in 2%SDS and TRIS. Standard concentrations were 0, .25, and .50 mg/ml of BSA. Froze lysate.
results: There was purple. The controls showed that putting in too much of the protein can reduce signal. 20ul set showed purple, the 2ul set didn't. after incubating for 30minutes.
yay it works.
562nm BCA test assay results
A very rough ball park of protein concentration of the whole cell is ~.1mg/ml (i think)
September
9-22-09
9-19-09
- outgrowth assay #4 with modified protocol:
modifications: 3 plates with negative controls on each plate, added 5ul of espA protein in 90ul of TRIS, incubated for 5hours, took out induced cells and put them in fridge for 6.5hours
- The results for the 9-19-09 outgrowth assay is here: results for outgrowth assay
It seems that the no pass constructs grow much faster than the needle constructs, and the OD measurements do not show significant different between the needle and no pass constructs. Although, when compared to the gliadin negative controls, the needle constructs did have a significantly higher signal.
new and improved outgrowth assay (includes BCA assay protocol)
9-16-09
- Outgrowth assay attempt #3. repeating.
changes made from the previous protocol:
1. incubated in 100ul total of espA (5ul espA protein and 95ul TRIS ph7.5)
2. incubate for ~1.75hrs
3. (washed 2X) blocked with BSA for 1.25hrs
concern: the film that covered the plate had pores, wasnt sure if when smoothing the film with hand w/out gloves caused any contamination.
Results: 9-16-09 outgrowth data
somethings to do next time: take OD right after adding media, use nonFITC conjugated espA in case the FITC has a negative effect on binding.
9-14-09
- Outgrowth assay attempt #2. looking kinda good.
Refer to the previous protocol, but with the following changes:
1. induced cells for 12hours
2. incubate maxisorp plate with espA for 1hour
3. added 100ul of 1mg/ml BSA in TRIS and incubated for 30min
4. added 5ul of cells with 95ul of 1mg/ml BSA in TRIS and incubated for 2hours
5. aspirated out cells and washed plate 3X
results:
looked at after 2.5hours, no growth
looked at after 6hours, growth, more than neg control: cl02365, ehab, oprF, hia Tecan data for 9-14-09
looked at after 7hours: growth, cl02365, ehab, azo
looked at after 8hours: growth, cl02365, ehab, upaG short, azo
9-12-09
- Outgrowth assay: protocol for 9-12-09 experiment
results: 9-12-09 outgrowth assay
9-11-09
- Ran the pull down experiment after inducing for 13hrs and incubating in espA-FITC for 8hrs.
The results are here from the experiment done on 9-09-09.
The results are not helpful because the wells with no cells had lower readings (so maybe the proteins stuck to the bottom of the plate?).
=9-08-09
9-8-09 pull down assay 1st run data
This is the first run of the pull down assay. This assay assumes that the cells will bind to a certain amount of FITC-labeled espA and when centrifuged, will be pulled down, leaving the supernatant with less fluorophore.
The results are not very telling. And there are some things that need to be improved.
Conditions for this experiment include: overnight inoculation to saturation. 5hr induction of 1:10 dilution of cells. incubation of cells with espA-FITC (1000X diluted) for 1.5hours. A titration curve was generated.
Improvements: need better aspirating technique for taking up supernatant. need to establish centrifuge speed and time. need a better titration curve.
9-07-09
results for assay: crappy - should maybe test the lysate. histag scFv. - know for sure that binding of espA and scFv is good? according to a paper - are you making protein period? tag with GFP. or functional assay (fail so far). increase volume to a larger scale system. run stuff on a gel preinduction and postinduction. or even do a western. - functionality period? test if in cytosol. lyse cells, do soluble binding assay. - if is functional, why is it not going on surface?? other ways to display. - what is it is functional but not functional on surface? add linker. test different linkers. other than linkers what other schemes can you do? is it N or C terminally fused. - buffers, salts, need something else to be active, sterics (like cells binding to eachother?), protein could have non specific binder but not likely. Tecan reading? What about the cell junk?
9-7-09 data
changes made from last time: incubation is 4hours and Tris buffer is used instead of PBS. A 1:100dilution and growth for 8hours is done in addition to 5hr induction
9-05-09
results for the assay
There was no significant readings for the needle scFv.
could it be: the induction (induce longer), amount of protein (add 10ul instead), the buffer (10XPBS), the affinity of the scFv to the protein (maybe too weak, so 3 washes washed all out), experimental design (the design does not give meaningful results in this assay), incubation time could also be too short (next time incubate for 4hours), a different buffer can be used (Tris instead of PBS, and pH it to 7.5), there might be an optimal pH for scFv activity
the pH of the 10X pbs used is 7.12.
9-4-09
- FITC conjugated the proteins: We did a spot test after pipetting the proteins out of the dialysis bag. There was protein. The protein was at exactly the pH in the protocol (which can be found in this notebook). Weighed out 3.8mgs of FITC. Added 300ul of the FITC solution into ~3ml of protein solution. Allowed incubation for 1hr at room temp. Put in dialysis bag, and will change solution tomorrow.
- Also grew up cells for the assay tomorrow. All 14 constructs and negative control is Gliadin scFv with cl02365 AtD. They were grown in 1ml of LB with AC antibiotics. They will be induced into triplicated tomorrow morning and allowed to grow for 5hrs before the assay.
9-3-09
- progress on needle assay: We purified the protein today, and it is dialyzing overnight. Tomorrow, we will FITC conjugate and grow cells in preparation for the soluble binding assay on Saturday.
- ran cellulase assay: data
-note that the cells were induced for over 24hrs, and allowed to sit for ~1hr on the bench top. The pH of the precipitation buffer is ~5, and the cells were spun down for 10minutes.
-There are definite color differences between the supernatants of the various constructs, and some of them were visibly very blue.
9-1-09
- cloning espA into histag vector:
PCR espA analytical gel -

expected size is 580bp
gel purified vector -

expected sizes: T7RNAP = 2383/4326bp (NcoI/EcoRI)
restriction map of espA in his tag vector -

expected sizes: 4326/580bp (NcoI/EcoRI)
sequencing - used ca56. no results b/c primer didn't bind (?)
Perhaps he did not sequence with ca56?
could the T7RNAP and T4lig have gotten mixed?
contamination on the plates, picked bad colony?
the restriction map gel looked good.
August
8-30-09
the two plates both look contaminated since there were large and small colonies. 3 clones from each plate was picked and will be restriction mapped tomorrow.
-should find out the part in the original his tag vector
- mgfp all in DH10B experiment: There are a lot of smears on the plate. AIDA does form a biofilm on the plastic and it can also be inferred that AIDA expresses on the surface of the cells. The mgfp-AIDA under the microscope seems to show a higher density of cells than the AIDA.
Should repeat experiment with the individual wells to prevent cross contaminating since we can't tell if some of the cells in the negative control are from the AIDA/mgfp or are background. The rfp film from previous exp. did not show up, so probably just strain (TG1 vs DH10B)
The cells from the DH10B exp: AIDA flocculated
- ran experiment with tyrosinase: pictures from the well plates are on the B144 computer and are in the iGEM2009 folder. We took pictures after each wash for 2 washes. Results are ambivalent as the negative seem to also have cells on the plastic well. ascorbic acid helped.
Not sure what is wrong - does tyrosinase need to be under certain conditions? incubation longer? pH not at 7? the buffer not phosphate but salt water? cells are expressing mgfp on the surface?
8-29-09
cloned espA into his tag vector. will pick colonies tomorrow.
8-28-09
- tyrosinase experiment. protocol used
result: no significant coomassie staining in the petri dish experiment. no negative control present either.
proposed protocol for mgfp tyrosinase exp
8-27-09
- ran experiment to determine whether AIDA adhered to the plastic on its own. have five samples: mgfp, mgfp with rfp, rfp, 1363, and AIDA with no passengers. I did two resuspension volumes, 50 and 250ul. The protocol for the petri dish experiments is on this page.
The results showed that there were spots of cells in the mgfp, rfp, and AIDA rows. Not as much cells were observed in the mgfp w/rfp.
It should be noted that the AIDA, mgfp w/rfp, and the rfp were in TG1 cells while the mgfp and 1363 were in DH10B cells. The negative control did not show visible films of cells.
- also ran same experiment but with 96well plate for separation. But the results seemed to show cells in both negative and AIDA, but not as much in mgfp (should look at again)
-will do experiment again with all samples in the right strain of cells DH10B. prep cells today.
-will do experiment with tyrosinase, will need to grow up all the constructs of mgfp again and also search the literature for some tyrosinase assays.
8-26-09
- run assay with rfp to determine if it will allow better visualization of cells under the microscope
-will run normal petri dish experiment: result- the cells were pretty clear (red) under the microscope, but both the negative control and samples had red cells.
- run assay with no passenger AIDA and construct (mgfp with rfp)to determine if AIDA has a role in the stickiness
-will run normal petri dish experiment: result- cells were again seen in both the sample and negative control
--perhaps the cells were washed off and stuck to other places. so either a 96well plate (with small surface area) or a large plate will be better for running the experiment. Also, the method of washing should be improved. Flick instead of rock?
mgfp-rfp and rfp only cells are TG1 and no pass AIDA are in DH10B
- flow cell update: might meet with Prof Herr tomorrow: will meet at 12pm
8-25-09
updated version of 8-14 sol gliadin assay data
8-24-09
- transformed just rfp into TG-1 cells to serve as a negative control: will pick colonies tomorrow, grow (pick from the other plate too) and do fiddling experiments on wed.
- induced cells today, so need to come back later ~8-9pm to try experiment?
- John will have IILK and mgfp stuff ready for experiment tomorrow.
8-21-09
- set of gliadin functional assay
- FITC conjugated
two sets of data that seem to indicate a signal for espP beta displayer:
8-14
8-15
however, a third set of data gathered did not confirm the trend.
- insoluble gliadin
a growth related assay where cells bind to insoluble gliadin in maxisorp plate was done and the results can be viewed here.
There was however, no difference in ODs after 7hours of incubation. so the assay is again inconclusive as to whether the cell's gliadin scFv is on the surface or functional.
- mgfp5 strep assay
strep data of mgfp5 and also other passengers
The results were unclear and the positive control is still considerably higher than the constructs.
- no passenger constructs strep data
raw data as well as comparison of linker vs no linker (INP)
- mgfp5 functional assays (fiddling)
We did several experiments with mgfp on a macro scale to test whether the cells stick to various surfaces, and whether they are able to set underwater.
-Spot test (an ASTM standard) - we scrapped a dried spot of cells that were previously on plastic petri dishes. The test showed that the spot was left a residue in the induced mgfps that were not present in the uninduced and 1363.
-sand mixed with bacteria and glass beads mixed with bacteria - we allowed the sand and bacteria to settle, to perhaps allow the bacteria to bind to these particles and make a cement like adhesive. However, our results have been inconclusive and there is no significant aggregating
It could be that the bacteria is coating the beads, but since they do not adhere to each other, there are no aggregation clumps formed.
-sticking toothpicks, pipet tips, and glass slides together - we also used the cells to stick these items together. They all stuck, including the negative controls. There might be slight differences in the adhesion, but since the test is qualitative, it can not be definitively proven.
-Washers and glass slides underwater - We conducted a test where we coated washers and glass slides with bacteria and then clamped them. We then unclamped the two pieces and watched for any noticeable adhesion. There was no observable adhesion in either the glass or metal case.
- spiking in water - an aliquot of cells were put on the petri dish and water was added to the dish before the cells dried to test whether the liquid would set. The test showed that there was diffusion and it did not set.
- other ideas for testing mgfp:
-laser tweezer
-flow cell
-midlog cells
- We did a coomassie stain on the midlog cells, and saw that the difference in phenotype (blueness) was higher with the saturated cells. Pictures can be found on Terry's camera.
Also looked at the glass beads under the microscope. Did not observed significant differences between negative control and construct. Also stained the beads with coomassie to try to stain cells that could be bound to the beads. The pictures of this are in a folder "igem 09" on the B144 computer. There did not seem to be much cells on the surface of the beads.
- co-transformed mgfp and rfp. The rfp plasmid is called pBjh1600-Bca1144.
colonies should be picked tomorrow.
- picked colonies of the mgfp and linkers Eco/Bam transfers. will need to be taken out of the fridge Sat.
8-12-09
- will run strep assay on 2 sets of mgfp5 and also perform the functional glass slide assay on the constructs
- will make the FITC conjugated gliadin in preparation for the gliadin functional assay. Today, remember to prepare the media and cells for the gliadin functional assay.
the temporary protocol for preparing the FITC conjugated gliadin is here
8-09-09
- Strep Assay of INP and Cel6A: INP showed signficant fluorescence for several autotranporter systems. There were also noticeable film of cells at the bottom of the 96-well plate that did not resuspend. The cells could be autoaggregating. For cel6A, most of the constructs had fluorescence readings close to the negative control. Also, there was observable whitish cell junk after resuspension in almost all of the wells with induced constructs. The uninduced and controls did not have this white cell gunk.
The fluorescence of the INP could be due to its smaller size. Linkers are a good subject to look into.
- strep assay data and graphs from all 9 passengers from the first set of parts:
cellulase data
ScFvs, Leucine Zippers, and INP
From the first set, only the leucine zippers and INP showed any significant fluorescence. Though the scFvs showed readings above the negative control, they were not fluorescent compared to the positive control.
The functional assays should be performed to better evaluate the passengers.
- UV pictures of the pellets for the strep assay from the 1st set of constructs:
File:UV pics from the first set.ppt
- re-inoculated all of set 1 from the working stock plates
8-08-09
- assembly of mgfp5 part: miniprepped 1st clone of 2-1, 2-2, 2-4, 3-3, 3-4, and Eco/Bam digested. Jenn will assemble these today.
![]()
expected sizes are: 1701/3171bp. The order is 2-1, 2-2, 2-4, 3-3, 3-4.
- Strep Assay: running EILD, KILR, Cel5B, and Cel9A.
remember to wash 96well blocks tomorrow for the assays
8-07-09
- assembly of mgfp5 part: All five clones came back perfect, so 3 colonies were picked and grown to be miniprepped tomorrow.
- Strep Assay (first run with pBca9495 and DH10B):
Induced the cultures that Jenn had grown up last night for 5 hours. Note that the positive controls pBca9145-9494 and pBca9495-9494 have 'Amp' and 'AK' resistance, respectively.
Three plates were assayed - cel3A, gliadin scFv, and needle scFV.
The results will be posted soon.
- some notes: when doing the 5hr inducing step, also include 1 or 2 uninduced constructs and the uninduced positive control.
- some notes: when doing the 5hr inducing step, also include 1 or 2 uninduced constructs and the uninduced positive control.
If strep assay will be run the next day, re-inoculate the constructs and controls the day before.
- specific notes about the 8-7 assay: the ODs were still ~0.1-0.2, but at least the positive controls also have similar ODs. There was pellet loss after the first spin down after strep-RE incubation. This occurred in the cel3A and gliadin plate, but not the scFv plate. (cel3A and gliadin plate - same wells where cell junk occurred) There was also some white insoluble material in the plates (cell junk?). (cel3A plate - A1-3 and 12, B1-3 and12, C1-3 and 12, D1-2 and 4, E1-2 and 4, F1-2 and 4; gliadin plate - D4, E4, F4) We did 2 washes only because of the pellet loss. The positive control is still very bright, and the fluorescence readings are a lot higher than that of the constructs. When compared to the induced negative control, however, the induced constructs seem to be higher (perhaps 1.5-2 times). The induced constructs are also noticeably higher than the uninduced constructs as well as the uninduced + and - controls. 4 different bottles of PBS were used.
- specific notes about the 8-7 assay: the ODs were still ~0.1-0.2, but at least the positive controls also have similar ODs. There was pellet loss after the first spin down after strep-RE incubation. This occurred in the cel3A and gliadin plate, but not the scFv plate. (cel3A and gliadin plate - same wells where cell junk occurred) There was also some white insoluble material in the plates (cell junk?). (cel3A plate - A1-3 and 12, B1-3 and12, C1-3 and 12, D1-2 and 4, E1-2 and 4, F1-2 and 4; gliadin plate - D4, E4, F4) We did 2 washes only because of the pellet loss. The positive control is still very bright, and the fluorescence readings are a lot higher than that of the constructs. When compared to the induced negative control, however, the induced constructs seem to be higher (perhaps 1.5-2 times). The induced constructs are also noticeably higher than the uninduced constructs as well as the uninduced + and - controls. 4 different bottles of PBS were used.
8-06-09
- assembly of the mgfp5 part: we miniprepped clones 2-1, 2-2, 2-3, 2-4, 3-3, 3-4 (these tubes are in the yellow box in Jenn's freezer), restriction mapped with Bgl/XhoI and Eco/Bam, and sent 2-1, 2-2, 2-4, 3-3, and 3-4 for sequencing. The results are in the sequencing log.

the first six lanes is the Bgl digest and the last six is Eco.
The expected sizes are: Bgl/XhoI = 10341bp and Eco/Bam = 1692/8649bp. The vector is pBdr052. The bands look correct except for clone 2-3 which does not have a second band for the Eco/Bam digest.
Sherine and I also Eco/Bam transferred the five clones and plated them. The cells were DH10B lefty cells.
- In preparation for the Strep assay (this time cells are DH10B and plasmid is pBca9495), we transformed 9494 and 1363 into DH10B cells, and put 1/2 of the rescue on plates and 1/2 into liquid culture.
8-05-09
- assembly of the mgfp5 part: picked colonies from plates, "new clone2" and "new clone3". There were few colonies from each plate. Picked 4 colonies and spot checked them on AKC plates.
will miniprep, restriction map, check for co-transform, and send for sequencing tomorrow.
- miniprepped the second set of E/B transfers, digested BseRI/XhoI and Eco/XhoI. Will run gel tomorrow.
8-04-09
- miniprepped and restriction mapped the mgfp5 basic part (in pir righty). there does not seem to be co-transformation, but will check one more time tomorrow morning for growth on spec plates.

lanes 1-4 are bamHI/XhoI digested. and lanes 6-9 are BglII/XhoI digested.
The proper sizes (for pBdr052) are 6654/2721 for BglII/XhoI and 9375 for BamHI/XhoI.
The bands look right.
Sherine did assembly of clones 2 and 3 from the "new" and plated. Will pick colonies tomorrow ~12pm.
- The internal passengers set of E/C plates were picked and colonies can be plasmid prepped early tomorrow morning and then restriction mapped.
8-03-09
- assembly by hand of mgfp5
- transformed into pir righty and plated. picked colonies and spot check on spec early 8/4. did two in parallel. the plate that is re-transformed had many more colonies than the one from the basic assembly.
- transformed into pir righty and plated. picked colonies and spot check on spec early 8/4. did two in parallel. the plate that is re-transformed had many more colonies than the one from the basic assembly.
- Eco/Bam transferred the internal passengers along with displayers
- digested vectors pBca9495KA-1144#5 and pBca9495AC-1144#5
- digested vectors pBca9495KA-1144#5 and pBca9495AC-1144#5

KA and AC. rfp~900bp and backbone~3kb
- i requested the split protein parts from the registry
8-02-09
- the plates of the Eco/Bam transfer of 16displayers looked contaminated b/c there were large and small colonies. Reasons for contamination...poor technique, Righty cells, the rescue media. Need to check, rescue media, vectors that the part digests were ligated into, the analytical gels of the digested parts (john).
despite that, I picked colonies and will miniprep and restriction map them tomorrow. Enzymes: BseRI and XhoI.
8-01-09
- picked colonies of the KA and AC pBca9495 plasmids and other random parts. Should be put into fridge tomorrow (or miniprepped)
- Eco/Bam transferred, and transformed 16displayers into righty cells. will pick colonies tomorrow morning. This is to prepare for final assembly.
July
7-29-09
- digested pBca9495 vector backbones for Eco/Bam transfer of the intermediate parts into pBca9495
The two gels are identical


Refer to Sherine's notebook for the reference to the lanes.
7-28-09
- ran strep assay on cel5B, cel9A, gliadin, and needle scFv.
File:7-28-09 strep assay of cel5B cel9A gliadin and needle.ppt powerpoint with UV pics and some OD normalized data (which were not very accurate because of the false positives from the normalization). The toxicity of the cells resulted in very low ODs and when normalized, the autofluorescence (which are about the same for all the cells) per cell seems to be higher. So data should just be analyzed by subtracting the background (LB)[next time, should have wells with just LB when take the OD, and subtract the OD from the LB]. And then check fluorescence with just PBS and subtract the fluorescence from that of the PBS only. Also subtract by the negative control.
The fluorescence data subtracted by the fluorescence of PBS. Also, subtracted from the negative control.
fluorescence data
From this data, some parts showed slightly positive signals:
cel5B - none
cel9A - VtaA11 AtD and Hag AtD
gliadin - azo1653 AtD, VtaA11 AtD, Hag AtD, Pcryo_1225 AtD, upaG_short, CPG_L2, ehaB, eCPX, yuaQ
needle - azo1653 AtD, cl02365 AtD (maybe?), CPG_L2
7-26-09
- Strep Assay of Cellulases, scFvs, and SOD
- Growth Assay of Cellulases, scFvs, and SOD
For details of the Strep assay on all cellulase parts: media:7-26-09_strep_assays_of_cellulase_parts.ppt
7-21-09
- Sequencing results from composite parts C09ig0021, C09ig0022, and C09ig0023. Looks good.
7-20-09
- miniprepped and restriction mapped four composite parts in pir-RIGHTY cells. C09ig0019 = cub w/term, 0020 = nub w/term, 0021 = caspace w/term, 0022 = Tev N w/term, and 0023 = Tev C w/term.

BglII:C09ig0019-0023 ladder BamHI:C09ig0019-0023
There is the right number of bands in each lane.
C09ig0019(6162/2837) C09ig0020(6162/2846) C09ig0021(6162/3533) C09ig0022(6162/3059) C09ig0023(6162/3077) ladder C09ig0019(8999) C09ig0020(9008) C09ig0021(9695) C09ig0022(9221) C09ig0023(9239)
The bands look like the right sizes. C09ig0021, C09ig0022, and C09ig0023 were submitted for sequencing.
- put C09ig0024-C09ig0040 set of parts ({<azo1653 AtD>}{dblterm}, etc) into pir-righty cells. They were accidentally put in pir lefty. Transformed and grew in 24-well block. (ordered down a column)
7-19-09
- transformed A2, B4, C1, and D1 as well as four other constructs from Jenn, into lefty cells. This tests the lefty cells to see if they are methylating correctly.
- Jenn did some more restriction mapping with Bam/XhoI.
7-18-09
- BamHI/XhoI restriction mapping of all basic parts (in assembly vectors). The expected sizes can be found on the google docs page: iGEM09 Parts

A1-H1

A2-H2 and A4-H4

A5-H5

A6-H6 and A7-H7
The goal of this restriction map is to determine whether the basic parts were in the correct righty/lefty cells. And also, can possibly be used to determine correctness of basic parts. Most of the bands look right, with a few that are ambiguous. For more details, see iGEM09 Parts.
7-17-09
- Colony pcr of 4 parts pbad, vta, ehab, and virG

oligos ca998 and G00101 were used. pbad, vta, and ehab were the right sizes but virG was not. The pbad were from a different clone from the ones used in assembly of the rest of the set. pbad and other parts will be sequenced (?).
- Colony pcr of pbad clone used for assembly

The pbad clone is not correct, could be reason for assembly failure, but need understand the bands.
7-15-09
- learned about assembly from Jenn
- looked at split fluorescent protein systems for the signal transduction project. Will find parts of these protein systems.
7-14-09
- researched various cellulases and cbms that could be made in the second set of parts. These parts with the DNA sequences are in a google spreadsheet: iGEM 09 Goal parts - Round 2.
Most of these cellulases are endoglucanases. Some of these enzymes can breakdown insoluble cellulose, and the rest break down soluble cellulose. Some of the other enzymes that degrade the plant cell wall (other than cellulose)were not included in the set, but can also be included. (xylanases, hemicellulases, and ligninases)
- need to search for good assays for: cellulose binding, cellulase catalytic activity, catalytic activity against other types of matter in plant cell walls.
- thought of an assay scheme that uses modularity. First individually assay for cellulose binding and for catalytic activity. Then find the best activity and binding to put in one system (bacteria) and assay again for catalytic activity. Objective is to increase catalytic measurements. Also assay for synergisms between cellulases. To add on another layer, should also assay hemicellulases and ligninases for catalytic activity and then combine the cellulases with these enzymes for improved activity against more complex plant matter. Also thought about the two cell system, where cells express different enzymes that will come at different stages to optimize the plant cell wall break down process.
- learned about gateway from Madhvi
7-9-09
- I picked colonies for ig213 and ig239.
Restriction mapping:
sent for sequencing?
- We took measurements of the first strep assay with varying concentrations of strep. There were 13 constructs from class. The results showed that all constructs had strep binding, which is not consistent with either the class or the
From the cells that were re-inoculated this morning, we did another strep assay, following a slightly different protocol.
The results are posted in the group meeting powerpoint Media:7_9_09 presentation slides.ppt.
It showed that the strep assay had good results.
7-8-09
- The forward oligo of ig213 and reverse oligo of ig239 were tube switched. John did another PCR with the correct oligos.
The gel purification picture of the pcr products showed the right size bands.
The product was then gel purified. The gp-d products are in the freezer. The two parts were cloned and plated.
- Gaby also did a PCA and a pcr to reassemble and amplify ig213 and ig239. For the amplification reaction, all four oligos were put in.
The picture of the analytical gel showed similar results as the pcr that John did.

ladder ig213 ig239
We stored the zymo cleaned pcr products in the freezer.
- We also re-inoculated cells and followed the Tecan assay protocol to do the Tecan assay. We noticed that there were a lot of pipetting error and also there is an extra transfer from the block to the flat bottom plate.
The constructs assayed were from the passenger displayer systems made in Bioe140L.
7-6-09
- Did colony pcr on the plates from pcr#4.

First three lanes are ig213 and last four are ig239. We used the specific forward and reverse oligos to pcr these parts, so if the wrong part was in the cells, then there should not be any bands. However, the gel showed that there are bands, meaning that the primers anneal, and some part was amplified. This could have ruled out our initial thought that these products were tube switched.
However, the bands could also indicate that perhaps the primers annealed to a region in the middle of the part, or that the part is repetitive and the pca assembly made a longer part (although im not sure about this).
- ran a gel on the colony pcr of ig239 that Madhvi did last night.

All of the lanes are clones of ig239. They all seem to have the right product, which is around 260bp. However, the bands could also be not the product. There are also two bands that are more faint than the other ones.
We miniprepped the four colonies (1, 2, 21, and 23) and and sent in two of them for sequencing. We will restriction map the others tomorrow.
- The spec LB was also contaminated. We are not sure when that happened.
- thinking about doing the PCA assembly and amplification again, except this time, starting from scratch. but will not change annealing temp and will not add DMSO. If doesn't work, should just keep re-amplifying.
7-5-09
- digested, gel purified (see gel here), ligated and plated ig213 and 239 from pcr round#3.
- John and I made spec plates.
- sequencing results for ig261, ig239, and ig314: perfect, perfect partial, and parent vector, respectively.
- Madhvi helped to do a colony pcr on ig239 from the 2nd round of pcr.
7-4-09
- I miniprepped the cells grown from 7/2/09 and restriction mapped them.

ig213 239 ladder 261 314
each part has two lanes except 261.
There are no bands except for ig314, which has a band very close to the size of the vector backbone, hence the brighter single bands in the last two lanes.

ig239 and ig261. each part has 3 lanes.
There are no bands for 239, but bands of the right size for all three of ig261.
ig239, 261, and 314 were sent off for sequencing.
- PCR#4 for ig213 and 239. with no DMSO. annealing temperature was 55C. started from amplification step and then did a reassembly. The gels for both steps are shown.

ladder ig213 239
There is only a faint band for ig239.

ladder ig213 ig239
There is only a faint band after the reassembly and amplification step.
7-3-09
- picked colonies for ig171 and 322 (re-pcr); and for ig213, 239, 261, and 314 (1st pcr)
- ligate and plated ig239 and ig261 (re-pcr)
picked colonies and grew cells up for restriction mapping and sequencing
- re-innoculated cells for Tecan fluorescence assay. The cells used were DH10B, TG-1, and MC1061. The plasmid used was -9494.
- re-pcr(#3) ig213 and ig239
did re-amplification step of PCA. The analytical gel showed no bands.

ig213 ig239 ladder
compared to the original analytical gel:

ig213 ig239 ig249 ig261
There are no bands for ig213 and ig239, but the pcr product was eluted with 8ul of water to concentrate the DNA in the zymo cleanup and faint bands were subsequently gel purified.
so we are did a re-assembly according to the PCA protocol and also did another amplification.

ladder ig213 ig239
The bands for each are of the wrong size. (ig213= 860bp, ig239= 260bp). Interstingly, they would be the right size if the two lanes were switched. However, that possibility is unlikely.
We zymo cleaned the pcr product.
- I tried to do the re-amplification step of the reaction once more. (pcr#4)
7-2-09
- Sequencing results from samples sent 7/1/09: successful parts - ig113, ig121, ig145, ig302.
- for re-pcr of ig213 and ig261, ran analytical gel, zymo cleaned, digested, gel purified.
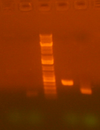
ig213 ig239 ig261 ig314
Only ig239 and ig261 had bands of the right size. ig239 has a dimmer band.

ig213 ig239 ig261 ig314
Only bands from ig239 and ig261 were cut out and gel purified.
- miniprepped and restriction mapped another set of cultures from the colonies picked on 7/1/09
restriction map of colonies from 6/27/09 plates

ig57 ig213 ig239 ig261 ig314
only ig57 showed a band of the correct size.
restriction map of colonies from 6/30/09 plates

ig79 ig137 ig171. each has 2 lanes.
All parts had at least one band of the right size.
samples were sent for sequencing.
- we also had a group meeting to update everyone on the progress of things.
7-1-09
- picked colonies from plates 6/30/09 plates ig79, ig113, ig121, ig137, ig145, ig171, ig322. (re-pcr)
miniprepped these samples ~9hours after picking.
restriction mapped samples from the new plates:



ig79 ig121 ig137 ig145 ig171 ig322
Each sample had a band at about the correct size. sent for sequencing, except ig322.
- miniprepeed cultures grown on 6/30/09. liquid from three tubes disappeared (perhaps dumped into waste or into other tubes).
restriction mapping of these parts:

0. ladder 1. ig121 ig137 ig261 ig239
2 lanes each with the exception of ig137 and ig239.
ig113 and ig121 showed bands of the correct sizes, while ig137, 261, and 239 showed no bands.

ig27 ig57 ig79 ig145 ig213 ig302 ig314
2 lanes each with the exception of ig79.
ig27, 57, and 302 showed bands of the correct size, while ig79, 145, and 213 showed no bands and ig314 showed a band that could be either vector or the insert b/c size is so similar.
sent ig27, 57, ig113, ig121, ig302 for sequencing.
- re-pcr of ig231, 239, 261, and 314 because these parts either showed no bands in colony pcr and resctriction mapping, or have had wrong sequencing results.
- re-picked colonies from 6/27/09 plates: ig27, 57, 113, 213, 239, 261, 302, and 314.
JUNE
6-30-09
- sequencing results for: ig1, 27, 57, 79, 113, 129, 137, 145, 171, 201, 207, 213, 239, 261, 314, 318, and 322.
There were five parts that are perfect. Some parts had very bad reads. Some had mixed peaks. Some had hairpins. And some samples did not sequence very long. This might be because the DNA is not pure enough, meaning that it could have had excessive salts, proteins, or EtOH.
Since the reads showed hairpins, should check the basic part DNA for these secondary structures. The mixed peaks could have been because of secondary structures or having more than one template.
- the 24-well block belly flopped in the shaker, so all of our cultures were discarded because of cross-contamination. :(
We re-grew cultures for restriction mapping and sequencing tomorrow. ig27, 57, 79, 113, 121, 137, 171, 213, 239, 261, 302, 314, and 322.
- Ran an analytical gel on the pcr products: ig79, 113, 121, 129, 137, 145, 171, 201, 207, and 322.

All the products are of the correct size.
The pcr products were then zymo cleaned, digested, and gel purified. Ig113, 121, and 137 were eluted with 6ul instead of 10ul because isopropanol was not added. Also 2ul instead of 1ul was added for those parts in the ligation. We transformed and plated the cells.

1. 79 2. 113 3. 121 4. 137 5. ladder 6. 145 7. 171 8. 322
6-29-09
- miniprepped samples: ig1, 27, 57, 79, 113, 129, 137, 145, 171, 201, 207, 213, 239, 261, 314, 318, and 322. and sent for sequencing.
- did colony pcr again picking from the 6/27/09 plates: 79, 113, 121, 137, 171, 213, 239, 261, 302, 314, and 322.

These bands were all under 500bp. ig113, 121, 137, 239, 261. each has 3 lanes.

These bands were larger than 500bp. ig79, 171, 213, 302, 314, 322. each has 3 lanes.
comments: restriction digests yields better results, so will do that instead of colony pcr. Re-pcr some parts because they consistently showed negative results on the gels.
grew up cells in 24-well block.
- PCR-ed: 79, 113, 121, 137, 145, 171, 322.
Did pcr again for ig322 and ig171 because these showed negative results consistently on the colony pcr gels.
6-28-09
- Colony PCR of ig1, 27, 57, 79, 113, 129, 137, 145, 171, 201, 207, 213, 239, 261, 314, 318, and 322.
3 colonies were picked from each plate

1. 113-1 2. 113-2 3. 113-3 4. 129-1 5. 129-2 6. 129-3 7. 201-1 8. 201-2 9. 201-3 10. ladder 11. 207-1 12. 207-2 13. 207-3 14. 137-1 15. 137-2 16. 137-3 17. 239-1 18. 239-2 19. 239-3 20. ladder 21. 261-1 22. 261-2 23. 261-3

1. 1-1 2. 1-2 3. 1-3 4. 27-1 5. 27-2 6. 27-3 7. 57-1 8. 57-2 9. 57-3 10. ladder 11. 79-1 12. 79-2 13. 79-3 14. 171-1 15. 171-2 16. 171-3

1. 145-1 2. 145-2 3. 145-3 4. 213-1 5. 213-2 6. 213-3 7. ladder 8. 314-1 9. 314-2 10. 314-3

1. ladder 2. 318-1 3. 318-2 4. 318-3

1. ladder 2. 322-1 3. 322-2 4. 322-3
The cells were grown overnight in 4ml spec LB: 1_2, 27_2, 57_1, 79_1, 113_1, 129_1, 171_3, 201_1, 207_1, 137_2, 145_3, 213_2, 239_3, 261_1, 314_1, 318_1, 322_2
- re-transformed and plated ig121, which we forgot about.
6-27-09
- sequencing: ig1-bad read, ig171-other sequence (other matched), ig249-perfect, ig310-perfect, ig318- one pt mutation at 220 (forward file) changing E to V and one pt mutation at 217 (reverse file). Can't decipher the middle part of sequence because ig318 is 1608bp.
For more information on sequence results, go to sequence analysis.
- Gaby and I re-transformed: ig1, 27, 57, 79, 113, 129, 137, 145, 171, 201, 207, 213, 239, 261, 314, 318, 322.
In the ligation, vector digested on 6/22, which were digested once, were used for ig201, 207, and 213. Vector digested on 6/27 was used to re-ligate ig 314, 318, and 322. The rest were ligated with the re-digested vector (6/26). The inserts were gel purified digested pcr products (there is only one tube of each).
TG-1 cells were used for the transformation, 2YT was used for the rescue, and 75ul of cells were plated.
- ig302-1, 306-2, and 322-2 were miniprepped and sent for sequencing.
- vector pBca9523-1144#5 was digested and gel-purified.
- stored in freezer box 2:
6 tubes of vector (10ul each)
miniprepped ig302-1, 306-2, and 322-2
- to do:
put the miniprepped DNA of ig249 and ig310 on stock plates.
6-26-09
- Small colonies of DH10B cells grew on both the spec plates from 327lab and B144, so the DH10B cells had spec resistance. We threw out all those cells.
- We also looked at the re-transformed ig302, 306, and 322. The colonies were large, round, and white. There was no parent vector bleed through. It could be that the concentration of vector we used was too dilute after re-digesting it.
We did colony PCR for ig302, 306, and 322. Ten colonies were picked from 302 and 306, and five were picked from 322.

1.-5. 302_1 - 302_5 6.ladder 7.-11. 302_6 - 302_10
The size of the pcr product is 1457bp. The parent vector part is 910bp. The bands are around 1500bp, indicating the correct product. Lanes 3 and 9 had no bands.

1.-8. 306_1 - 306_8 9.ladder 10. 306_9 11. 306_10 12.-16. 322_1 - 322_5
The sizes of ig306 and ig322 are 898bp and 2821, respectively. There are bands for all ig306 lanes. Although the length of ig306 is really close to vector part length(910bp), the colonies we picked were not pink, so the bands should still correspond to the correct product. There were no bands in the first two lanes for ig322, and two faint bands (both under 1kb) were present in the last three lanes. No product of the correct size was found for ig322.
We re-inoculated cell cultures for 302_1, 306_2, 322_2, and 322_3 to grow them up for sequencing.
- We miniprepped the cultures from block "L" and block "R" using the Machery-Nagel Miniprep protocol. A restriction digest was done for restriction mapping.

1. ig1 2. ig27 3. ig57 4. ig79 5. ig113 6. ig129 7. ladder 8. ig137 9. ig145 10. ig171 11. ig201
The bright bands above 1.5kb are most likely the vector backbone (2472bp). Of the lanes that showed two bands, lanes 1 and 10 seem to have the correct bands.

1. ig207 2. ig213 3. ig239 4. ig249 5. ig261 6. ig302 7. ladder 8. ig306 9. ig310 10. ig314 11. ig318 12. ig322
The bright bands above 1.5kb are most likely the vector backbone. Lanes 4, 9, and 11 have bands that approx. correspond to the sizes of the correct products.
ig1, ig171, ig249, ig310, and ig318 were submitted for sequencing.
- Gaby and I miniprepped the cells that contained pBca9523-1144#5, that Jenn had re-transformed.
- tubes put into the freezer box:
miniprepped ig1, ig171, ig249, ig310, ig318
two tubes of 50ul of miniprepped vector pBca9523-1144#5
- to do:
miniprep 302_1, 306_2, 322_2, and 322_3 for sequencing.
analyze ig1, 171, 249, 310, and 318 sequences.
re-transform.
6-25-09
- We looked at plates for ig1, 27, 57, 79, 113, 121, 129, 137, 145, 171, 201, 207, 213, 239, 249, 261, 302, 306, 310, 314, 318, and 322.
There were large and small colonies, indicating contamination. There were also many pink colonies, indicating parent vector bleed through.
Sources of contamination could be: DH10B cells, old EtOH, non-sterile technique, or other.
- We found that the LB and plates from B144 were not contaminated. To test the DH10B cells, we grew the competent cells on spec, amp, kan, and chloramphenicol plates. We also replaced the ethanol and cleaned our benchtops.
- We re-digested vector Pbca9523-1144#. The vector had been previously digested with EcoRI and BamHI, but it is thought that the vector might not have completely digested (vector backbone after digestion is 2472bp and the entire plasmid is 3382bp).

0. ladder 1. vector 2. vector 3. vector
We then re-transformed the ligated products of ig302, 306, 314, and 322 into TG-1 competent cells instead of DH10B cells. For the transformation, we used 30ul KCM instead of 20ul. The glass spreader was used instead of the wire loop spreader.
- Colonies were also picked from the plates and will be grown overnight in two 24-well blocks.
block "L"
| 1 | 57 | 113 | 129 | 145 | 201 |
| 1 | 57 | 113 | 129 | 145 | 201 |
| 27 | 79 | 306 | 137 | 171 | 207 |
| 27 | 79 | 306 | 137 | 171 | 207 |
block "R"
| 213 | 239 | 261 | 302 | 318 | 314 |
| 213 | 249 | 261 | 310 | 318 | 322 |
| 239 | 249 | 302 | 310 | 314 | 322 |
| - | - | - | - | - | - |
note: there were no colonies on plate ig121.
- Elicia transformed the 10 basic pcr constructs into TG-1 cells.
- Jenn will transform vector Pbca9523-1144#5 using the stock tube from Chris, and we will have more vectors for ligation.
- We talked about using google spreadsheets to keep track of all our tubes in the freezer and also use color coded labels (for example, digested vector vs undigested vector). Also, in general, we should organize the tubes and keep all our work areas as sterile as possible. Running controls to test for contamination is also good.
- John made a good resource for SOEing procedures.
- to do:
check DH10B test plates for colonies
check the plates of TG-1 cells
miniprep and restriction map the innoculated colonies
pick colonies from the TG-1 plates
re-transform ig121(?)
6-24-09
- analytical gel of PCR#2 product

1. 302 2. 310 3. 314 4. 318 5. 322 6. ladder
note: lane 3 (ig314) has a band at the wrong length and lane 5 (ig322) does not have a band.
ig302, 310, and 318 were zymo cleanedup, digested using the thermocycler program "37C60min", gel purified, ligated, and transformed. 70ul of cells were plated, and the plates were stored in incubation on the 3rd floor. To test for contamination, the LBs used for rescue were plated. An empty spec plate was also incubated.
note: for plating: new LB and spec plates from 3rd floor were used. Also, an insert previously known to ligate into our vector was used as a positive control
- Redo SOEing PCR for ig314 and ig322 using pcr protocol. changes: used 4K55 program instead of 2K55 and pcr machine with two blocks.
analytical gel of PCR#2 for ig314 and ig322

0. ladder 1. 314 2. 322
The pcr gave correct sized bands.
The procedures and protocols used were the same as above and ig314 and ig322 were transformed and plated.
- gel purification of PCR#1 for ig306

0. ladder 1. 306A 2. 306B
note: TG-1 was used as template for pcr instead of MG1655. The two previous PCRs failed because we used the wrong template.
The analytical gel after the SOEing pcr showed a band of the correct size.

1. 306 2. ladder
The procedures and protocols used were the same as above and ig306 was then transformed and plated. In the ligation, a vector with the same backbone but different insert was used. New ddH2O and LB were used for plating.
- We also checked to see how well our colonies grew from the first 12 plates. Only 57A, 113B, and 129B grew. But not sure if these are right.
6-23-09
Checked plates from the first set. colonies were small. Next time, should do a negative control with just parent vector. Before picking colonies, note color of the colonies; they should not be red. There were many red colonies and some contamination. Not sure of source of contamination. (LB? Plates?)
24 well block for inoculation of cells from plates of the first set.
| 1A | 1B | 113A | 113B | 145A | 145B |
| 27A | 27B | 121A | 121B | 171A | 171B |
| 57A | 57B | 129A | 129B | 201A | 201B |
| 79A | 79B | 137A | 137B | 207A | 207B |
A= small colonies
B= big colonies
note: A&B of 57 and 129 are both small colonies since the large ones were pink.
used LB with spec antibiotics b/c of vector.
will grow up cells and do restriction mapping to verify parts.
Second set of oligos came in!
Gaby, John, and I will be doing the SOEing constructions.
Patrick and Sherine - PCA assembly
Joey, Elicia, and Tom - basic PCR constructions
SOEing construction of second set of parts
| name | {<part>} | oligo name | plate location |
| B09ig302 | {<virG AtD>} | O09ig302/O09ig301 O09ig300/O09ig303 |
C1/B1 A1/D1 |
| B09ig306 | {<yuaQ AtD> | O09ig306/O09ig305 O09ig304/O09ig307 |
G1/F1 E1/H1 |
| B09ig310 | {<AIDA-1 AtD>} | O09ig310/O09ig309 O09ig308/O09ig311 |
C2/B2 A2/D2 |
| B09ig314 | {<cel3A!} | O09ig314/O09ig313 O09ig312/O09ig315 |
G2/F2 E2/H2 |
| B09ig318 | {<cel5B!} | O09ig318/O09ig317 O09ig316/O09ig319 |
C3/B3 A3/D3 |
| B09ig322 | {<cel9A!} | O09ig322/O09ig321 O09ig320/O09ig323 |
G3/F3 E3/H3 |
Plate with oligos is for both basic PCR and SOEing constructions is labeled "Plate B09ig277 10uM"
PCR#1 - removal of restriction sites using cloning by PCR. PCR programs used were 4K55 for ig314 and 2K55 for the rest.
Gel purified the products of PCR#1 following the SOEing PCR protocol

0. ladder
1. 302A
2. 306A
3. 310A
4. 314A
5. 318A
6. 322A
7. 302B
8. 306B
9. 310B
10. 314B
11. 318B
12. 322B
note: lanes 2 and 8 did not have bands. pcr for ig306 failed, so will re-PCR.
Set up PCR#2 - for amplification of part with restriction sites removed. follow same protocol as first pcr.
re-PCR ig306 in same pot as PCR#2.
will do: zymo cleanup of PCR#2, gel purify ig306, restriction mapping of first set cells.
stored in yellow box 2:
PCR#1 products in pcr tubes labeled "A302" etc.
gel purified PCR#1 products in epp tubes labeled "ig302A gp-soeing 6/23", "ig302B gp-soeing 6/23"etc.
6-22-09
Ran analytical gel of the first set (12 PCR products) of the PCA assembly.

0. 1kb ladder plus
1. ig57
2. ig79
3. ig113
4. ig121
5. ig129
6. ig137
7. ig145
8. ig171
9. ig201
10. ig207
11. ig1
12. ig27
12 for 12!
Lanes 8 and 9 were very faint. These parts were small (under 200bp), and so were lower in the gel.
The list of basic part lengths (approx. since DNA were not digested) can be found in the construction files of the respective basic parts.
Digestion of vector pBca9523-Bca1144#5 and of first set pcr products.
Followed EcoRI/BamHI Digest of vector plasmid protocol and EcoRI/BamHI Digest of PCR Products protocol. The pcr program used is "ecobamdi" under the iGEM folder.
Gel purification of the 12 products

1. vector
2. vector
3. ig129
4. ig137
5. ig145
6. ig171
7. ig201
8. ig207
9. ladder
Lanes 3, 4, 7, and 8 showed no bands. These were under 300bp, and probably ran off the gel. ig129, 137, 201, and 207 will be digested again.

1.ig1
2. ig27
3. ig57
4. ig79
5. ig113
6. ig121
7. vector
8. ladder
Patrick and Sherine melted the gels and did a zymo cleanup of the pcr products and the vector.
Gaby and I redigested ig129, 137, 201, 207, and vector pBca9523-Bca1144#5 following EcoRI/BamHI Digest of PCR Products and EcoRI/BamHI Digest of vector plasmid.

1. vector
2. vector
3. vector
4. ig207
5. ig201
6. ig137
7. ig129
8. 1kb ladder plus
Gel purified with zymo cleanup. The ig parts were eluted with 8ul water and the vector was eluted with 10ul water. See Sherine's notebook for other modifications to the protocol.
Ran ligation reaction following EcoRI/BamHI Digest of PCR Products protocol. See Sherine's notebook for modification to the protocol.
Transformation by heat shock of plasmids into DH10B cells. Used 70ul instead of 75ul of cells.
6-19-09
PCA assembly of first set of parts
| PCA | |
| name | {<part>} |
| B09ig57 | {<OprF AtD>} |
| B09ig79 | {<cl02365 AtD>} |
| B09ig113 | {<VtaA11 AtD>} |
| B09ig121 | {<Hag AtD>} |
| B09ig129 | {<Pcryo_1225 AtD>} |
| B09ig137 | {<Hia AtD>} |
| B09ig145 | {<type IIIs Needle Complex scFV>} |
| B09ig171 | {<gliadin binding scFv>} |
| B09ig201 | {<Cub>} |
| B09ig207 | {<Nub>} |
- finished up to the zymo cleanup of the amplification step. stored all tubes in yellow box in freezer.
- stock oligo mixture (100uM) in pcr tubes labeled "R, F, mix, ig57" etc.
- cleaned up assembly labeled "ig57 6/19 tem" etc.
- diluted forward and reverse oligos from 100uM to 10uM. diluted oligos labeled "F57" and "R57" etc.
- cleaned up amplified parts labeled "ig57 6/19 amplified" etc.
Followed PCA gene synthesis protocol to assemble and amplify gene sequences. The PCR programs used were PCA1 for assembling oligos and PCA2 for amplification; both are in the iGEM folder. Zymo cleanups were done after both assembly and amplification steps.
cloning step of protocol still to be done.

{kind=link}
{kind=link}