Janet B. Matsen:Lab Tips & Tricks
Back to Janet
I came to the Lidstrom Lab with zero molecular biology or synthetic biology experience. I have great appreciation for all of the resources put online by the OpenWetWare community and other websites, so I am hopeful that I can share some of what I have learned. Please contact me with questions, comments, and corrections.
Cultures
Media
- TB (terrific broth) gives you healthier cells and more plasmids than LB. I use LB for freezer stocks because or media prep person makes it in abundance but TB for minipreps to get higher plasmid yield.
- SOB media ("super optimal broth") can be used like TB. The recipes are somewhat variable.
- SOC media is SOB with added glucose; it can be used when cells recover from transformation.
- Our lab and Rob Egbert haven't needed to use it as of 2012/10/3. But I am considering making some SOC for electroporation of Gibson assemblies.
- Adam Wargacki claims he recovers 2-5 times more colonies with SOC. They buy commercially prepared competent cells. As he moves to using less and less of the aliquots per transformation, he prefers to use SOC. He also suggests SOC for difficult to transform plasmids, such as 30kb+ plasmids, and even prolonging the recovery time to more than 1 hour. However, if you expect the transformation to work, there isn't much advantage over LB.
Pouring Plates
- Before melting agarose to pour, put it in a heater that keeps agar liquified for a few hours. This makes it melt much faster so you won't have to babysit it.
- Once melted, put it in an empty styrafoam shipping box so it won't develop solidified chunks. You can leave the lid off or leave it partly on to slow down the cooling.
Sterile Technique
Keep an extra clean set up pipette tips
- I find it useful to keep a super fresh box in the drawer that I only pull out for occasions when sterility is critical. When the less sterile tips on my bench are used up, I remove the sterile sticker from a drawer box and move it to the countertop.
- I like that it is inside my bench so nobody can accidentally open it under non-sterile conditions.

- If the not-so-sterile tip boxes accumulate, it is ok to autoclave our brand.
- Alert: Autoclaving (on the dry cycle) loosened the filters so they are not sufficiently held in place. Turning a tip upside down is sufficient to release a tip after autoclaving.
- Additionally, Andrew said they always autoclave filters used on the bioreactor in the wet cycle to prevent damage to the filter.
- I haven't tried to autoclave on the wet cycle yet (3/27/2013), but that may be more useful.

- They DO melt in the waste autoclave cycle (Casey the dishwasher said so 3/2013)
Contamination on plates
- Don't be surprised if you get contamination on plates with multiple antibiotic resistances. There are plenty of plasmids floating around natural systems with multiple on them already. Bacteria make antibiotics in natural systems to add competition to an an environment. (Mary Lidstrom, 2012)
Numbering for Experiments
- I find numbers more useful than labels for most of my experiments. Keep a master google doc spreadsheet. With these, you can do better documentation than a lab notebook and elaborate tape labels allow. example spreadsheet, example commentary for corresponding spreadsheet.
- This also saves time with labeling, and reduces the risk that you will mistake one sample for something with a similar name. It also provides an order for your samples at each step of handling. You can label plates as you make them, cultures as you inoculate them, etc.
- I can't stand the feeling of being confused about something I did experimentally in the past, and this is my best solution.

DNA Magic
Minipreps
Grow in TB, not LB
- This alone gives me order(s) of magnitude higher miniprep yields.
Grow overnight cultures with sufficient head space
- the air volume should be at least 4 times the liquid volume.
- increasing the culture volume seems like a good idea at first but the lower oxygen concentrations are detrimental to plasmid yields.
- Specific suggestions for amounts of culture grow pSB plasmids overnight before miniprepping:
- pSB1A3 (high copy number): 2mL of TB is sufficient for miniprepping. 5 mL in a 15 mL test tube is great.
- pSB3K3 (medium copy number): 5+ mL in green-capped test tubes (they are 25+ mL)
- pSB4C5: I have only done pSB4C5 (low copy number) once I prepped from 50 mL TB (overnight in a 250 mL flask) and got a very pleasing 200 ng/uL in 50 uL. The absorbance at 230nm looked higher than usual but the DNA peak was good. Now I usually do 5-10 mL in a green-capped tube, but have never gotten 200 ng/uL!
The strain you use really matters!
- I do most of my minipreps from Top10 strains. If you use other strains, make sure they are cloning strains. Cloning strains have nucleases knocked out, making your miniprep more stable.
- In 10/2012 I tried miniprepping DNA from BL21(ai) and doing a restriction digest. The DNA was totally degraded afterward. I compared the phenotypes of Top10 and BL21(ai) using this list and found that Top10 has EndA1 knocked out, whereas BL21(ai) didn't.
- Elizabeth "Betsy" Skovran taught Amanda to use PB buffer. The composition is proprietary. We use this on our Thermo Scientific miniprep kit: add 500 uL after loading DNA.
PCR
- Optimizing PCR rxns:
- Try the PCR with 55oC for annealing and 0% DMSO first, almost always. If that doesn't work, you will see the manual gives you MANY options for things to change including template concentration, primer concentration, annealing temperature, Mg2+ concentrations, polymerase concentration, alternate buffers, extension time, and annealing temperature. There is no way you could test all the combinatorial possibilities here. I generally only change the annealing temperature and % DMSO if my template concentration is within the suggested range.
- When Tm = 55oC failed, I used to do 12 rxns (20 uL each) with 4 temperatures (on a gradient PCR block), each at 4 temperatures ranging from 55oC - 70oC, and 0%, 5%, and 10% DMSO. If one of these conditions don't work, there is little hope that changing other things will be a game changer. Now that feels like too much work & I only do a few temps w/o DMSO.
- Try the PCR with 55oC for annealing and 0% DMSO first, almost always. If that doesn't work, you will see the manual gives you MANY options for things to change including template concentration, primer concentration, annealing temperature, Mg2+ concentrations, polymerase concentration, alternate buffers, extension time, and annealing temperature. There is no way you could test all the combinatorial possibilities here. I generally only change the annealing temperature and % DMSO if my template concentration is within the suggested range.
- If you are unsure whether you added an ingredient to the PCR mix, add it again.
- I *always* have a written list of components and the volume I want to add and I pen in a dot next to ingredients I have added. However, it is possible that you can get distracted and be unsure whether you added a component. If that component is template, nucleotides, or enzyme, I always add it again. I have never been unsure about whether I added buffer or DMSO (when desired) so I can't promise performance if you double these concentrations.
- I also add the most abundant & inexpensive components to the mix first. Thus, I add water, buffer, then primers. If I am unsure that something is wrong at this point, I can scrap it without any lost of scarce or valuable materials.
- If making more than one master mix, label the tubes of master mix by writing the number of the primers on the lid. Then you can easily double check that the right primer goes in the right mix as you create it.
- I don't actually do this because I like only having 1 kit. Keeps things simple.
- Annealing @ 55oC in PCR before anything else often works great. You are more likely to get undesired bands due to mis-priming but it is often a great place to start.
Gels
- You can re-use tips when loading into wells if you rinse in the TB buffer in your block between every step. This is great because tips are ~$40/box. Don't do this for important samples (just in case.)
- If you are gel-purifying large volumes of PCR product, you can tape the lanes together. The greatest dangers are (a) the well ripping and allowing your sample to leak out and (b) having the buffer slosh you sample out of the well when you move or bump the box. Come talk to me about extremes in taping because I have made all the possible mistakes.
- I don't actually do this any more (2012/07/02) because I am using Gibson cloning and don't need much DNA any more.
- Running large volumes on a gel ("overloading" the gel) can cause the band size to run too fast or too slow. If you are running a large amount for purification, include a lane next to the ladder that is not overloaded so you can be confident you know the band size. The following gel ran alright, but a slight shift is visible.

- If you have a voltage source that will stop after a set amount of time, feel free to run your gel as you leave and image it in the morning. Diffusion of bands shouldn't be a big problem unless they are << 500 bp.
- Don't do this if you are going to cut & gel purify the band.

Purifying DNA
- purifying DNA on a gel column has ~ 90% loss. purifying DNA on a column (non-gel) has ~ 90% recovery. avoid gel purification. (Justin)
- Freeze 'N Squeeze Gel extraction may be worth a try.
- I'd like to try. I even have enough of a kit to try some, but I never have samples I feel like taking the risk on! The only person I know who does it (also the person who gave me a piece of a kit) swears by it.
- Purify gels on gel-specific kits. Purify PCR products on PCR product specific kits if you have one. The former does a less effective job removing primer dimers. (Justin)
Eluting DNA from miniprep & gel extraction columns
- By default, most people elute from miniprep and gel purification columns in water instead of the supplied buffer. This is motivated by the desire to have DNA that is free of buffers and other compounds that may inhibit subsequent reactions (e.g. PCR or Gibson Assembly). You may, however, suffer from lower recovery of DNA from your column as the pH of the water you use is likely < 7.5.
- Both the Qiagen gel extraction kit and Thermo Scientific miniprep kit use 10 mM Tris·Cl , pH 8.5 elution buffer.
- Should not inhibit PCR. (No EDTA, a chelator)
- Gibson assembly mix is ~ 200 mM Tris, pH 7.5. Adding a few uL of 10 mM pH 8.5 Tris shouldn't have a huge effect. I'd like to call their tech support and make sure, however.
- Is ligation the main issue? I'm not doing a lot of restriction cloning right now, so this wouldn't be a huge issue for me. (10/2012)
Equipment
Rebound board for ergonomic pipette dispoal
- Check out my rebound board for discarding used tips! I won't be shooting tips onto Amanda's bench any more.

Ice Buckets
- the best type of ice bucket we have in lab are the ones that perishable reagents are delivered in when mailed. These are more insulating and have excellent lids. They are also useful to use when flash freezing samples with liquid nitrogen. I have no idea why nobody else has these in their starting lineup.
An ode to scotch and boxing tape
- Masking tape and boxing tape are extremely great when laid on top of markings on your tubes. This will make your labeling more resilient to ethanol exposure and mechanical wear. It helps preserve your writing when your rub off ice that accumulates on the top when tubes are stored at -80oC. I cover any labeled tube that I will be keeping for a while with tape.
- Warning: I have gotten eppendorf tubes really stuck in a centrifuge before when I use cryogenic labels and overlaid them with boxing tape. I was able to get them out easily without working hard by putting the rotor in a 60oC oven for a while before pulling on the tubes with plyers, but this should be avoided.

Transformations
When plating transformations you expect will work
- If plating a transformation of miniprepped DNA, you can do two things to save time/mess:
- Plate with wooden spreading sticks
- If you expect decent efficiency, there is no need to use roller beads, a flame sterilized stick, or any other extra tool.
- Just use a pipette tip
- Move the cells to the plate with a pipette tip (works for ~20 uL, but may not be as great for 200 uL)
- bend the pipette tip by pressing it carefully against the sterile side of the plate lid
- Spread cells
Nonunifrom plating is good for miniprepped plasmids you expect to transform with reasonable efficiency
- I like nonuniform spreading because it will allow you to grab single colonies even if your transformation efficiency is really high or really low.
- If the efficiency is high and you plate it nice and evenly, you can have a lawn across the whole plate making it hard to pick single colonies. If the efficiency is low, you don't need to distribute it evenly anyway because a handful of colonies in a relatively small area of a plate will still give you single colonies you can pick.
- I plate gibson assemblies (usually low efficiency) with beads.
Outgrowth over the weekend is not recommended
- Don't transform plates at room temp over the weekend, especially if you rely on Amp resistance. Often you see a lot of contamination!
Electroporation is MUCH MUCH more efficient than chemical transformation
- Transformations done with 1uL of 0.1 ng/uL DNA (pGA 3K3 RFP from Rob Egbert). 0.1 ng is ~10^8 plasmid copies. Tami's electrocomp cells yielded several hundred colonies, which suggests the transformation efficiency is on the order of 100/10^8 = 0.000001 = 0.0001% efficiency. The chemically competent cells yielded < 10 colonies, which is much lower efficiency.
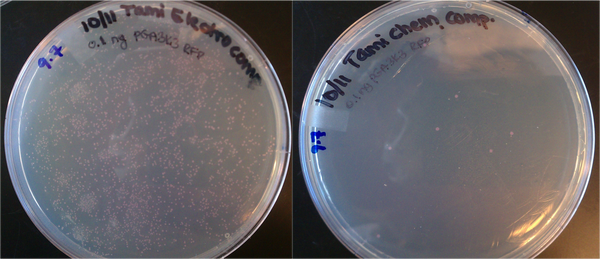
Top10 cells prepared for electroporation and chemical transformation. Both were transformed with ~10^8 copies of DNA (0.1 ng of plasmid.)
Make your electrocompetent cells super viscous
- Really concentrated cells perform better; my electrocompetent cells are so viscous they barely pipette. The picture below is with more dilute electrocompetent cells that I currently (8/2013) use but illustrates the importance of concentrated cells.

- My cells clump into a "booger" during recovery (pre-plating) that is mostly unspreadable. The clumping is less of an issue if you don't centrifuge them before plating. For this reason I recover in 200 uL and plate all of it.
Keeping primers, plasmids, strain stocks, & sequencing organized
- Keep a master google spreadsheet with a page each for primers, plasmids, strain stocks, and sequencing. (example) This will keep you very organized, and allow you to collaborate more easily.
- On the primers page, each primer should have an individual number, a name without spaces, the sequence, comments for its intended use, and the date you ordered it. When ordering primers from IDT, you can paste the three consecutive columns with primer number, name, & sequence into the multiple primer entry window. Then delete the space between the primer number & name, and add a hyphen. You can grey out primers you decide are no longer useful (example: you find out it is wrong or doesn't work).
- On the plasmids page...
- On the strain stocks page...
- Keep the primers, plasmid stocks, cell stocks, and strain stocks in boxes with their unique identification number on top. Order them by number. This allows for ease of sharing resources & information. Note that the plasmids can be stored as minipreps and in the cloning strains, so you will have two boxes that share the same numbers.
- Keep a dropbox folder with the final versions of all of the plasmids you trust & keep.
- I name them by their plasmid number & short description.
- In this, I keep an extra copy of the sequence file that is annotate with all of the confirming sequencing rxns I did. For each sequence that comes back, I add a feature that is named with the order number of the sequencing reaction & the primer that was used to obtain the sequence. The feature should cover only the part of the sequence for which the sequencing reaction was solid. This means visually trimming off areas with N's.
Buffers
pH depends on dilution of a buffer and presence of salts
I wrote two sections here:
- Why does the pH of a buffer change with dilution?
- Why does the pH of a buffer change with the addition of salts (changes in ionic strength)?
Software
Using BLAST
- Useful intro for beginners and intermediate users: Chapter 16: The BLAST Sequence Analysis Tool
- Alignment metrics:
| Metric Abbreviation | Metric | Basic Description |
| bit score | bit score | calculated from a formula that takes into account the alignment of similar or identical residues, as well as any gaps introduced to align the sequences. Can be compared across searches (it is normalized). |
| E-value | expect value | indication of the statistical significance of a given pairwise alignment and reflects the size of the database and the scoring system used. The lower the E-value, the more significant the hit. A sequence alignment that has an E-value of 0.05 means that this similarity has a 5 in 100 (1 in 20) chance of occurring by chance alone. Might not be biologically significant though! |
- Legend for alignments:
| Alignment Feature | Description |
| plus | Conservative substitutions (that is, the substitution of amino acids whose side chains have similar biochemical properties), as judged by the substitution matrix, are indicated with +. Note: it doesn't stick to the groupings we are taught in biochem. I have seen Q (-ONH2: polar, uncharged) paired with K (NH3+: charged), Q paired w/ E (- charged COO-), N (uncharged -ONH2) paired with + charged H (N ring), etc. Haven't found anything that summarizes what they consider "conservative". 9/24/2013. Is it only separating hydrophobic from all others? No. D and P aren't marked w/ a +. |
| dashes | insertions or deletions |
| lowercase gray letters | low complexity |
| X | Amino acid residues in the query sequence that have been masked because of low complexity are replaced by Xs |
Uncertainty Codes: As you work with nucleic acid sequence records and read scientific articles, you may come across representations of bases which are not A, T, G, or C. In fact there are several of these in Figure 3.3. These nonstandard letters are actually the uncertainty codes proposed by the International Union of Biochemistry and Molecular Biology (IUBMB) to represent certain groupings of nucleotides. On occasion, software or a scientist is unable to decide if a newly sequenced base is, for example, an A or a G. The three IUBMB symbols most frequently encountered are R for the puRines (A or G), Y for the pYrimidines (C or T), and N for aNy nucleotide. Here are the uncertainty codes:
| IUBMB symbol | Definition |
| R | A or G |
| Y | C or T |
| K | G or T |
| M | A or C |
| W | A or T |
| S | C or G |
| B | C or G or T |
| D | A or G or T |
| V | A or C or G |
| H | A or C or T |
| N | G or A or T or C |
Using APE (free plasmid editing software)
- Pros:
- Free --> can have it on all of your computers
- Allows annotation libraries --> Easy annotation of new plasmids
- About the annotation libraries:
- This is one of the most useful tools. You can edit them in any text editor. To use them, use the drop-down menus to load the library, then click to annotate your current sequence using this library.
- I keep an annotation library that holds all of my sequencing primers, and use this to sequence clones. I open my plasmid annotated with genes, etc., then load this library of primers and overlay the primers on top of my sequence. If you have annotations of the regions of your plasmid that you have sequence verification for, it makes it very easy to find primers that will sequence other regions you are interested in.
- Beware:
- When you are searching for a sequence in your APE file, it is possible that it will incorrectly tell you it is not there
- This is happening because the DNA is stored a linear, not circular. If you have the cursor clicked mid-way through the file, the "find next" button will only look in the portion of the sequence that is between your cursor and the last letter. This can be avoided by storing all DNA as circular. There is a rectangular button in the upper right corner you can click until it says "circular."
- The alignment tool has given me ... unusual... results. Instead, I mostly use BLAST and use the blastn option.
- Note: FASTA format needs to have a space after the >
- When you are searching for a sequence in your APE file, it is possible that it will incorrectly tell you it is not there
Mendeley
- Mendeley : reference/paper manager that's awesome
- Janet's workflow: (1) discover papers via google scholar saved search, (2) save them to Mendeley in the appropriate nested folder regardless of whether you intend to read it now or "some day when I'm thinking about that topic" (3) read in Mendeley, which makes a green dot (indicating unread) go away.
- You can highlight and keep notes on top of your PDFs enabling quicker review of topics.
- Imagine being able to query all of the papers you have ever read for a topic of interest! You can also see whether you have ever read a paper or not, potentially saving yourself time.
- I also like the idea that I will never lose track of a paper I want to tell someone about. It isn't uncommon for someone to tell me about a paper but be unable to find it and send it to me. This should never happen for a dedicated Mendeley user.
- I'm not thrilled with the word plug-in for inserting references.
Shipping Samples
Shipping samples containing proteins on dry ice
- Beware that shipping samples on dry ice (solid CO2) can affect the pH of the sample. It probably doesn't matter for RNA seq samples, but I know some of us send samples containing protein on dry ice from time to time.
- evidence: http://www.nature.com/nmeth/journal/v10/n4/pdf/nmeth.2409.pdf
- "Proteins generally exhibit low solubility near their isoelectric point (pI). Therefore, acidic proteins have an increased tendency to aggregate or precipitate as pH falls below physiological levels."
- Interestingly, a pH change did not occur when they stored the samples in a -70oC freezer for 72 hours. The CO2 had time to dissipate. Note the tubes are CO2 permeable!
- evidence: http://www.nature.com/nmeth/journal/v10/n4/pdf/nmeth.2409.pdf
Tech Support
I have a lot of questions, always. The longer I work with a piece of equipment or technique, the more I get into the nitty gritty. Thus it is important for me to have good resources to answer the questions I come up with.
- It is incredible that in this day and age, manuals aren't available online for some manufacturers.
- 1/2013: I'm trying to use some of the manual integration features on a Shimadzu HPLC. These features aren't addressed in the manual. They don't make any of their manuals available online!
- If you can, buy an Agilent HPLC instead!
- 1/2013: I'm trying to use some of the manual integration features on a Shimadzu HPLC. These features aren't addressed in the manual. They don't make any of their manuals available online!
- I have the best things to say about BioRad and Agilent and would happily buy their products.
- Though they may not be the least expensive, but they make their manuals available on line and have lots of publications and tutorials for the basics relevant to their equipment.
- Biorad has excellent technical support. I have always been connected to helpful and insightful PhD holding individuals who have firsthand experience with the methods and tools I'm using.

{kind=link}