Janet B. Matsen:Guide to Gibson Assembly
Back to Janet
Hello! This is an *old* wiki page I wrote in grad school. I'm now a data scientist at Zymergen. See what I'm up to on Twitter and LinkedIn.
Intro
This tutorial is an aggregation of the lessons/tips/tricks I have learned while using Gibson cloning for dozens of diverse cloning projects. It is intended to supplement available protocols with some advice and warnings that I hope can save you time with your assemblies.

- Gibson assembly allows for seamless cloning, pretty easily.
- Assemblies are independent of sequence, and you are not restricted to use of restriction enzyme cut sites.
- Assemblies are scarless.
- You can assemble multiple pieces, from multiple DNA sources (plasmids, genomes, etc.)
- It can be used for site directed mutagenesis: NEB guide
- The efficiency drops as the assembly size increases (>8 kb starts to become a problem) and as the number of pieces increases (3-4 is ok, but I haven't tried more). It is also lower when cloning toxic genes.
Steps (concise)
- Design oligos to yield 20 - 100 bp overlapping linear DNA segments
- Clean DNA fragments (column cleanup, or gel if necessary)
- Use Gibson Assembly Mix (now commercially available)
- Transform
- Electroporation is usually used to provide higher yield.
- Screen w/ colony PCR
- Sequence 1-3 candidates
Other Resources
- Spreadsheet template I made to help with the Gibson workflow: spreadsheet template.
- You can duplicate it by signing into google, clicking on the link, and clicking File --> Make a Copy. -JM
- Benchling Gibson assembly tutorial. Benchling is a rad DNA editing tool, that is free for open-access DNA sequences.
- NEBuilder: A web-based primer design tool
- Guide by the creators of Gibthon (software I haven't tried)
Steps & Tips
See below for consolidated version.
Prepare plasmid maps
- Make a plasmid map of what your completed design should look like
- This is key. You will want it for primer design, checking your primers, assessing sequencing reactions, etc. I use APE, open-source software. See my APE use comments in Tips & Tricks.
- Mostly, this means copying from other plasmid sequences and pasting into a new plasmid file.
- However, you can add shorter items like promoters and ribosome binding sites by coding for them in your primers.
- You can also add longer regions of DNA using longer (90+ bp) oligos
- You can use genomic DNA, usually from whole cells (no need to purify first)
- Double-check your design
- Make sure each gene has a promoter, RBS, and stop codon if desired.
- If you aren't familiar with your sequences, make sure the sequence has no stop codons in frame with the start.
- Make a plasmid map (e.g. APE file) for each segment you will PCR amplify from a template (optional)
- Do include overlap generated by the primers.
Design primers
- Primer design is key. If all of your PCRs give good yield with no undesired bands at all thermocycling conditions tested, your assembly is very likely to go well.
- It is always a good sign when primers work at several annealing temperatures that are a few oC apart, and across DMSO concentrations. Since the assembly step is so dependent on primer sequence and absence of single stranded DNA structure (hairpins, etc.) the ease of PCR is a good indicator for whether the assembly is likely to go well.
- If your backbone doesn't amplify well, or amplifys with side products and requires gel purification, you are much less likely to get successful assemblies. If a poor PCR is generated, consider increasing the annealing temperature of the binding region for the primer > 72oC, and using different areas of the vector for overlap if possible.
- I always use primers with Tm >72oC for the annealing region. Tm values should always be calculated by the Finnzymes website
- This formula is applicable to Phusion DNA polymerase, the DNA polymerase used to create the DNA you will assemble.
- Tm = 80oC worked for me (11/2013) when I was doing site directed mutagenesis and wanted to confer more overlap.
- Make sure the reverse primers you are ordering are in fact reverse complemented.
- Use cheap primers. The primers should confer 20-100 bp of homology between to adjacent overlapping segments. 40 - 100 bp is ideal; substantially shorter or longer will give you lower yields.
- You usually only need one of the two primers to confer homology. If you use an 18-30 bp primer for one edge of a seam, and the other primer is 60 bp (including binding and homology), that is usually enough overlap.
- Keep in mind the pricing structure from the oligo company you use.
- If ordering with IDT, primers should usually be 60 bp if you are encoding homology.
- The price per base pair jumps when you add the 61st base pair: we pay ~$9 for a 60 bp primer but ~ $34 for a 61 bp primer. Using less than 60 bp reduces the length of the homology between adjacent DNA pieces in the assembly.
- Sometimes you need a longer (say 90bp) primer to add promoters/RBSs, or additions to a coding sequence. These primers work fine with the default purification; don't pay more for additional purification just because they are long.
- If ordering with IDT, primers should usually be 60 bp if you are encoding homology.
- Optional: Check primers for cross dimers with Finnzyme's multiple primer analyzer. If the annealing temperature of the primer dimer(s) is low, this will probably not be a problem during PCR.
- I don't do this any more.
Double Check your Design
- Make sure the forward primers and reverse primers you are ordering match the intended direction.
- This is an easy mistake to make.
- Fill out a table like the picture below so you have an explicit record of the assembly.
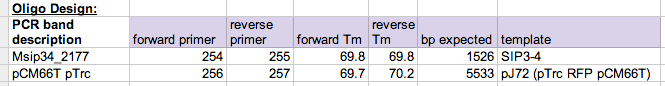
Start a record keeping spreadsheet that has the primers, Tm values, template used, and expected bp. Access to spreadsheet depicted: here - You can reference these cells when you plan out PCR reactions.
- You can blast your primers and templates with blastn to make sure they only anneal where you expect if you aren't super familiar with your sequences.
- You can blast the APE files for the expected PCR products against each other to make sure they have sufficient overlap.

Generate PCR fragments
Find optimal conditions
- I run each PCR at a few annealing temps and DMSO concentrations. Example below:
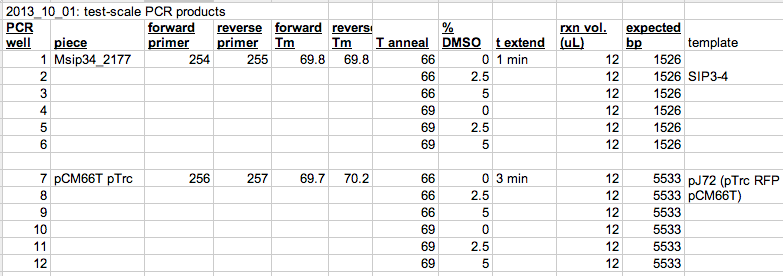
Example of test-scale DNA synthesis batches. Spreadsheet: here. - DMSO can be important, especially if you are amplifying DNA from the genome of whole bacterial cells. The DMSO likely disrupts the membrane enough to allow the polymerase to work.
- Run a few uL (~4uL) of each PCR product on a gel to identify rxn conditions that yield a lot of product. Look for conditions that make a lot of your product, and ideally no other undesirable products.
- Using Tm = 70oC (not lower) reduces the probability of unspecific bands greatly.

Dpn1 digestion of template DNA
- Dpn1 should usually be added after the PCR is complete to degrade the template DNA. This will reduce the number of background colonies when you transform. It is not usually necessary when gel purifying fragments, however, gel purification should be avoided as discussed herein.
- This is to avoid template carry-through.
- If the templates for your PCRs are Kanamycin vectors, and you are building a Kanamycin vector then some fraction of your transformants will just be cells with the template plasmid(s) carried through. This needs to be kept in mind later at the screening step.
- You can put 1/2-1 uL in your PCR product is complete; there is no need to modify the buffer first.
- You will only get background if the antibiotic marker of the template is that of your design goal.
- If you have a fragment from an Amp plasmid, and are building a Kanamycin vector, there is no need to add Dpn1.
- gel purification without doing Dpn1 digestion usually is sufficient to greatly reduce background.
- here is a sample result of background for a scenario where I used ~0.5 ng of template plasmid per 25 uL of PCR reaction to produce my backbone, then column purified (not gel purified!), and didn't do a Dpn1 digestion. The pink colonies are the plasmid template carrying through the column purification, into the assembly reaction and transformation step.
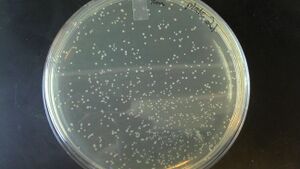
pink colonies in this transformation of a Gibson assembly are carry-through of template from the PCR amplification of the backbone
- This is to avoid template carry-through.
Warnings
- Be extra careful that you use the right combination of primers if you are amplifying multiple fragments from one plasmid, or if your primers work across templates used for an assembly.
- E.g.: once I was trimming a vector, and use the wrong combination of primers for the backbone. The band on the gel appeared correct (400 bp difference on a 5kb backbone is subtle) but lead to assemblies with only one of the two seams being correct.
Make enough to purify and assemble
- Run purification scale reactions to make DNA for assembly
- If your product is specific and doesn't need to be gel purified: (only needs PCR cleanup)
- 20uL of a strongly amplified insert is plenty. Do a bit more (30uL) if it is the backbone. These amounts usually yield plenty of DNA for 5+ assemblies, allowing the possibility for multiple attempts.
- If your product is co-amplified with other undesirable products and will need to be gel purified:
- run more like 60-120 uL, depending on how bad the byproducts are.
- If your product is specific and doesn't need to be gel purified: (only needs PCR cleanup)
Purify PCR fragments
- The best way to purify PCR products is a simple column cleanup. We use the Qiagen PCR cleanup kit, and elute in water.
- This usually requires your PCRs were very specific to the band size you wanted. This is why PCR primers are done with melting temperatures of 70oC: doing annealing at a high temp (67-70oC) is the most likely way to give you the desired PCR amplification. You need to have checked ~2-3uL of your product on an agarose gel to make sure your PCR was specific to your goals.
- You can also gel purify your PCR bands, but you lose a LOT of product, and the product is lower quality.
- This will remove primer dimers, and undesired bands. Unfortunately, the column-based gel extraction kits have extremely low efficiency. You can elute in water or the buffer provided by the kit (presuming it is only 10 mM Tris, pH 8.5 & has no EDTA), but I always used water.
- Elute in 30 uL (not 50 uL) to provide a concentrated product.
- Desired outcomes:
- Column purifying 30uL of a strong PCR band should yield ~40 uL of ~30-50 ng/uL product.
- Gel purifying ~100 uL of PCR product usually yield ~ 50 ng/uL.
- You will want ~ 60 ng of backbone in ~ 5 uL for assembly so concentrations as low as 12 ng/uL are usually fine.
Nano Drop PCR fragments
- Run 1.5 uL on a NanoDrop machine to approximate the DNA concentration of each eluate.
- You can use the 1.5uL run on the machine into an agarose gel to confirm that the products look good, and weren't accidentally mixed up during the purification.
- Example:

The 1.5uL aliquots of purified DNA that were used for NanoDrop were then run on a gel.

Gibson assembly reaction
- add your purified PCR products and add water to reach the desired concentration as specified by your commercial kit or home-brew recipe.
- The commercially available kit works ~10x better than some home-made mix in our lab.
- 50oC for 1 hour
- do in a thermocycler, and have it hold between 4 and 15oC forever afterward
Transformation
- Usually electroporation is used, as it can give you much higher efficiency. However, some people use chemically competent cells, which can be sufficient if the assembly is simpler. How much DNA you transform and whether you need to desalt it first depends on your expected assembly efficiency and product toxicity.
Desalting:
- Desalting DNA for 15 minutes on millipore filters means you can add more DNA to electroporations and not have arcing.
- We use Millipore desalting paper, item #VSWP01300 to desalt 12-20 uL reactions and Millipore # VSWP02500 for larger volumes. Put the whole assembly on top of a filter that is floating on top of water.
- Aaron Puri waits for 15 minutes of desalting, and electroporates at 1.6kV without arcing. -6/2015. There is no harm leaving them longer.
Electroporation:
- Can be much more efficient then chemically competent cells.
- Use ~3uL of assembly if the assembly was not desalted. The more assembly mix you add, the higher the salt concentration and the more likely your sample will arc. Arced samples have much lower viability but are still worth plating.
- 1-3 uL is usually plenty if you have a high efficiency at assembly.
- If you used the commercial assembly mix and your design (a) isn't too complicated (too many pieces, too big of a final product, too toxic of genes) and (b) is transformed into very good (concentrated) electrocompetent cells, then 1-2 uL may give you enough colonies to have a lawn.
- JM use Top10 E. coli. Electrocompetent cells need to be super concentrated. There should be only enough 10% glycerol in the mix to allow pipetting. If the viscous cell suspension gets a little bit stuck while you are pipetting, you are around the right viscosity. See Janet's notes
- To desalt & thereby be able to use more DNA per electroporation:
- We use Millipore desalting paper, item #VSWP01300 to desalt ~12 uL reactions and Millipore # VSWP02500 for larger volumes. Put the whole assembly on top of a filter that is floating on top of water. Leave for 1 hour. You can transform the whole reaction after this if you wish. A simple assembly (2 pieces, normal to small backbone & normal insert size) should give a lawn.
- You may want to transform some un-assembled fragments, to get a sense of what the background in the real transformation. (Some of the colonies that grow up on the transformation plate will have template DNA, not your desired construct, despite Dpn1 digestion.
Plating:
- If you are plating plasmids conferring Ampicillin resistance, plate on carbenicillin not ampicillin.
- Ampicillin is notorious for giving satellite colonies or even lawns of non-resistant bacteria. This is especially a problem if your assembled plasmid leads to slow growth, as the non-resistant bacteria will have plenty of time to flourish. If your electrocompetent cells are good, then the high cell density will likely lead to a lawn of bacteria on an Amp plate, even if most of the bacteria aren't Amp resistant. You could plate a small fraction of your electroporation on Amp, but that presumes you have a high assembly efficiency and a low-burden plasmid (e.g. mild promoter + RFP, not high strength promoter and multiple enzymes).

Screening: Colony PCR
Use colony PCR to generate PCR fragments that will confirm your assembly.
- Don't use Phusion for this; it is much too valuable.
- I use [2X OneTaq http://www.neb.com/nebecomm/products/productM0486.asp] PCR mix for several reasons:
- It is cheap
- It can be stored in the fridge, thawed, for months without harm.
- It has loading dye already so loading into agarose gels for observation is expedited.
- Tip: Make about 1/2 mL of water and colony PCR primers in appropriate proportions for the final 1x mix. See this spreadsheet for mix proportions. Go ahead and make 1/2 mL, you may be glad you have extra later and water/primers are almost free. You can store leftovers in the fridge indefinitely; I label mine OneTaq 212 213 if I mixed in primers 212 and 213. Then when you to do colony PCR, you can add 6 uL of this mix to each well, dissolve cells in each well, and add 6uL of the 2x mix on top.
- Alternately, you can make a 1x mix (add the necessary water and primers) and use the mix after many freeze-thaw cycles.
Colony PCR design:
- PCR over a region that is a different length than any of your template plasmids. This will allow you to tell which are successful assemblies and which are template carry-through.
- If you changed a gene in a plasmid, and the gene size is different, PCR for the length of this region.
- You can PCR across the whole insert if you inserted into an empty vector and your templates won't amplify to give the same product sizes.
- If you changed a promoter, chose a primer that only amplifies only if the new promoter is present.
- If you did something like site directed mutagenesis, colony PCR can't help you distinguish templates from successful assemblies.
- You can have more than two primers, such that each template and the desired product will yield a different size products, allowing you to see which template has carry-through.
- If you changed a gene in a plasmid, and the gene size is different, PCR for the length of this region.
- If you don't have any regions that have changed significantly in size (e.g. you are doing site-directed mutagenesis), it is best to have transformed some of the linear fragment products to get a sense for how much background (template) DNA is carried through. It is also extremely important to have done Dpn1 digestion in this case.
- Include the templates as controls, especially if using more than two primers.
To do colony PCR:
- You can decide to replate colonies you tested before or after your results are in. I always restreak once, aiming to get single colonies, to reduce the probability that my miniprep will be a mixed population.
- I plate at the same time I sample if:
- I think the fraction that are successful (not template) will be high
- I am running the PCR overnight and won't get the results until the morning.
- I don't plate until after the gel is run if:
- I think the results will be mostly carry-through of template plasmid
- I will have an opportunity to run the PCR products in a gel before I leave for the day, allowing me to only restreak "winners."
- I plate at the same time I sample if:
- Decide how many colonies you want to screen. Prepare a PCR strip (or strips) with the wells numbered and matching the colony numbers.
- I use set(s) of 12, because my agarose gels have enough lanes for this and two lanes of ladder. You will use at least one of the wells to amplify the template DNA as a control. I do more colonies (up to 33-34) if I expect template carry through to be an issue, or if the genes are toxic and successful assemblies make the cells unhealthy.
- If you are restreaking every colony that is tested, prepare your plates now. I divide the plate into 6 pie slice shapes.
- With a permanent pen: circle the colonies you want to test, and put numbers (e.g. 1-10) next to each. The numbers will allow you connect successful PCR reactions to successful colonies.
- If replating in the beginning, also mark the pie slice areas with these same numbers.
- Put 6 uL of water/primer mix in each well.
- After transformation, use a pipette tip to grab part of a single colony on a small pipette tip. It is best if you can see a little biomass on the tip, but you don't need/want much more than that.
- If you are restreaking colonies now: wipe a little bit of the colony on the plate, then dissolve the rest in the corresponding numbered PCR well.
- If you are not restreaking colonies now, try to leave some biomass on the plate, but be reassured there are always cells left unless you really punched a hole in the agarose.
- It is possible to overload it if you have really big colonies and suck up a lot of it with the pipette tip.
- Once a given PCR well has colony dissolved in it, eject the pipette tip into the well behind it. In addition to having each well numbered, and the colonies numbered and circled on the transformation plate, this is an added safeguard to ensure only one colony is put in each PCR reaction.
- Run the PCR with the correct extension temperature of the enzyme & the correct annealing temp for the primers. (68oC for OneTaq. 55oC works for VF2 and VR primers)
- Run the PCR products on a gel with ladder, such as Fermentas MassRuler
- If you haven't restreaked winners, do so at this point.
- Inoculate from a single colony into selective media the following day.
Sequencing
- Select 2-4 colonies for sequencing based on colony PCR
- Sequence the seams of the Gibson assembly first.
- Sequence the other regions, as it is possible a PCR error was introduced
- Usually when an "error" is found, it was actually present on the template.
Consolidated Version of Protocol
Note: I have prepped a spreadsheet template that may make your first Gibson experience easier. You can duplicate it by signing into google, clicking on the link, and clicking File --> Make a Copy. -JM
- Make a plasmid map of your design
- Design Primers & generate annotated sequences of the bands you intend to create
- primers should confer 40-100 bp of homology & be 60 bp long (in most cases)
- 62oC < Tm < 65oC as calculated by the Finnzymes website
- Check primers for cross dimers with Finnzyme's multiple primer analyzer
- Make sure the reverse primer is reverse complemented!
- Double check primer design before ordering.
- Blast your primers and templates with blastn and make sure they only anneal where you expect. If there is a potential for mispriming with a high (>55oC) annealing temperature, consider trying to alter your design to prevent problems during PCR.
- Blast the APE files for the expected PCR products against each other
- Generate PCR fragments
- Run each PCR with a few annealing temps and DMSO concentrations
- Check ~ 1.7 uL of each PCR product on an 0.7% agarose gel and identify reaction conditions that gave product and don't have undesired bands.
- Optional: the good DNA can be treated with Dpn1
- Use ~ 1 uL per 50 uL PCR product to degrade unwanted template DNA
- Purify PCR fragments
- Gel or sometimes PCR cleanup.
- Elute in ~30 uL to obtain a concentrated product.
- Measure DNA concentration with a NanoDrop system
- Gel or sometimes PCR cleanup.
- Plan Gibson Assembly reaction
- Use ~ 60 ng of backbone and stoichiometric quantities of insert(s)
- Transform
- Electroporate 1 uL into a cloning strain. Can do multiple electroporations and plate the cells together after they have grown out at 37oC.
If you get stuck
At the assembly step
- Make sure your bands are good, and aren't contaminated with undesirable bands. Run ~2uL of the DNA on an agarose gel.
- Tens of nanograms are visible.
- If you have short pieces, you can sew them together with overlap extension. It is often easy to sew two pieces together if one is short (<1kb) or if both are < 2-4 kb. Sewing together larger (~4kb) segments will probably cause you trouble. See Overlap Extension PCR
- You are more likely to get PCR errors incorporated if you use this method.
- You can make two assemblies that are each closer to your design goal, and reassemble them into the desired final product.
- This is handy when you have a large number of pieces (>3), and is particularly valuable when your design is large (9 or more kilobases) or your genes are toxic.
Time saving tips
- All components can be kept in the fridge for months without harm, enabling you to start PCRs in minutes. There is no need to spend time waiting for components to thaw, or putting them away at -20oC.
- Have a spreadsheet that it set up for streamlined workflows, with auto-referencing of cells.
- My example:
- Enter the components in the first page, with a picture of your sketch.
- Auto-calculates Phusion master mix solutions based on # of reactions, and max % DMSO
- Auto calculates amounts of DNA to add to Gibson Assembly mixes.
- Keep track of all PCR products, Gibson assemblies, and transformations.
- I use a PowerPoint document in parallel where I paste in screenshots of my work, including:
- PCR wells, and auto-calculated Phusion master mixes. Check off ingredients as you add to master mix.
- Photos of resulting PCR products run on gels.
- Commentary on decisions.
- In principle you could put the pictures in the google spreadsheet, however, I find this to be too busy. Here is a sample .ppt. It is not intended to tell a story so don't worry about any of the details. It mostly shows how I pair screenshots of my google docs with the gels, and add lots of annotations.
- My example:
Questions asked about the sample spreadsheet
- The numbers for the forward and reverse primer cell in the oligo design sheet what are they exactly?
- These are just unique numbers for each PCR well. It is best to be as organized as you can, because you never know when you need to re-do a PCR product or know what is inside of PCR strips that have been on your counter for a week or so. You can name your agarose gel images with these numbers as descriptions, and they can be used to identify conditions and context of PCR products you purify and assemble.
- Does the "bp expected" on page 1 include the overhangs?
- Yes, I Include the overhangs. This is recorded here because it is the size of the band you will be looking for on your agarose gel. Most products are big enough that you wouldn't be able to tell the difference between PCR products that differ by 40-80 base pairs, so it usually doesn't matter if you record this super accurately.
- I am having a little trouble following you DMSO% calculation.
- DMSO isn't added to the master mix, but the amount of DMSO you will use is relevant to how much water you add to the master mix. I add 1.2 uL of 50% DMSO to wells that contain a total volume of 12 uL. For the 0% DMSO and 5% DMSO wells, I add 1.2uL of water and 1.2uL of 25% DMSO. The box in the upper left, "", is for whether you want to have a max DMSO = 5% or 10%. I used to go up to 10% routinely but now I only go up to 5%. If the Tm of the annealing portion of your primers is really ~70oC then you don't usually get any benefit from added DMSO. In fact, added DMSO most often leads to no effect or prevention of PCR products from forming at all. So in summary, you only enter the percentage for the highest value you will go to, and use corresponding dilutions of DMSO in water to achieve the lower percentages.
Examples
- Break up backbone if it is large (> 4kb??)
- Only need 2 short primers to break it up: the homology is free.
- you can chose where the seam is if you use longer oligos
- RFP for backbone: don't screen red colonies!
Tricky Cases
- Replacing short sections like ribosome binding sites
- primer will necessarily have homology in two places. **DRAW SKETCH**
- HOMOLOGY
- Causes problems during PCR and assembly. Homology within a hundred or even a few hundred base pairs of the end can lead to recombination, as the exonuclease can be very fast.
- toxic protein
- if you are trying to clone in a toxic protein, your assembled plasmid may be too toxic to yield colonies
Making your own Gibson mix
- We used to make our own before New England Biolabs started selling it, but ours gives ~10x less colonies so we no longer make it.
- Tips:
- Balancing the ratio of T5 & Phusion is important given the mechanism. The exonuclease is so concentrated relative to the desired concentration in the mix that it should be diluted 10X before use.
Thanks
I want to give a special thank-you to Robert Egbert, who got me started with this technique in 2011 and changed the way I do research. (linkedin)
