Lidstrom:Electroporation
From OpenWetWare
Jump to navigationJump to search
Back to Protocols
- Electroporation is chosen when you need higher efficiency of transformation, or when you want to try transforming multiple plasmids at once.
- It is often difficult to get multiple plasmids in at once.
Electroporation is much more efficient than chemical transformation
- Transformations done with 1uL of 0.1 ng/uL DNA (pGA 3K3 RFP from Rob Egbert). 0.1 ng is ~10^8 plasmid copies. Tami's electrocomp cells yielded several hundred colonies, which suggests the transformation efficiency is on the order of 100/10^8 = 0.000001 = 0.0001% efficiency. The chemically competent cells yielded < 10 colonies, which is much lower efficiency.
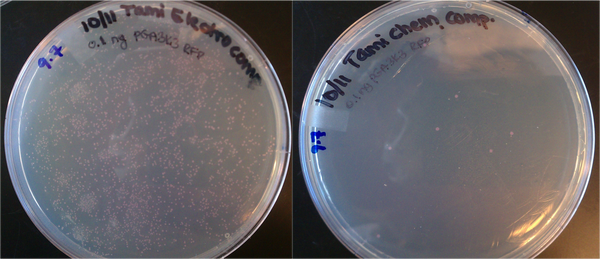
Top10 cells prepared for electroporation and chemical transformation. Both were transformed with ~10^8 copies of DNA (0.1 ng of plasmid.)
Preparing Electrocompetent Cells (in progress)
- More details are found here Competent Cell Preparation but the basic idea is as follows:
- Grow cells in LB (+ antibiotics, often)
- Consider using a "better" media than LB: one that contains sugars and Mg2+.
- Wash 2+ times with sterile, ice-cold 10% glycerol
- Resuspend in 10% glycerol
- Concentrate up to 500 times relative to culture volume.
- Aliquot into sterilized eppendorf tubes
- Flash freeze with liquid N2 and store at -80oC
- Grow cells in LB (+ antibiotics, often)
Preparing Cuvettes
Chosing Cuvettes
- We bought these: these (7/24/2012) but are likely to switch to these.
- Originally, we wanted autoclavable cuvettes, however, it seems that ethanol sterilization is effective.
Sterilizing cuvettes for reuse
The cuvettes should be washed for reuse. If washed properly, they can be reused up to 10 times. For each cuvette, do the following while waiting for the cells to recover:
- Squirt 95% ethanol to fill cuvette more than halfway. Add cap and invert cuvette a few times to disinfect it. Pour out in waste receptacle.
- Wash cuvette three times with DI H2O using squeeze bottle and dump water into sink.
- Squirt 95% ethanol in the cuvette, add the cap, and let it sit for an hour or overnight.
- Pour out ethanol and place in UV hood to dry.
- Store in freezer for next use
It seems that the cuvettes rust when left to soak in solution, increasing their rate of arcing. Note: most cuvettes are made from plastic that will melt. Do not autoclave.
- Don't leave them in a bleach solution. Don't leave them in water. They will rust, increasing the probability of arcing.

How much DNA do I use?
- Use 1-3 uL of Gibson assembly
- If you want to use more, do a PCR cleanup desalt to remove salts & prevent arcing.
- We use Millipore desalting paper, item #VSWP01300. Put the whole assembly on top of a filter that is floating on top of water. Leave for 1 hour. You can transform the whole reaction after this.
- If you want to use more, do a PCR cleanup desalt to remove salts & prevent arcing.
- plasmid miniprep: 100 pg (1/2 uL or less is usually sufficient)
- multiple plasmids at once: 5 ng each.
- We haven't had much success with this yet.
Transforming E. coli
- Add 1-10 μL of DNA source (see guide above) to the electrocompetent cells, swirl the tip gently in the culture to mix, and incubate on ice block for one minute.
- Working quickly, transfer the contents of the aliquot into the center of the gap of an ice-cold electrocuvette from the freezer, and then place the electrocuvette in the electroporator. Check that the electroporator is set to deliver 1.8 kV, and then press both of the orange buttons to deliver a pulse. The display will show "Ch 9" and eventually beep; release buttons when you hear this beep.
- Rob Egbert & the Klavins lab use 1.25 kV
- Again, working quickly, remove the electrocuvette from the electroporator and add 1/2 - 1 mL of room-temperature LB to it, pipetting up and down several times to extract the cells from the 1 mm gap.
- Transfer entire contents of electrocuvette back to the tube.
- Check and record the discharge time constant (units are in milliseconds).
- 4-<5 seconds is good. (Amanda noticed that with no cuvette, it read 5.3 sec. You might not be making good contact if you're over five seconds))
- Incubate tube transformed cells at 37°C for 30-60 minutes.
- Klavins lab does 30 - 45 min; our lab usually does 45 - 60 min.
- put plate in 37°C incubator with agar side up overnight
Alternate transformation protocol: when your DNA concentration is LOW
From Rob Egbert/Klavins Lab: Transformation of diluted competent cell aliquots for low-efficiency constructs
- This protocol can be necessary when performing oligo assembly or other high-complexity Gibson assembly reactions. The general idea is to purify the product of the Gibson assembly reaction using a PCR purification column, which removes enzymes and salts, and to transform the purified product in high volume (5 to 10 uL) to diluted cell aliquots.
- PCR Purification of Gibson assembly reaction
- Follow PCR purification protocol to purify the Gibson assembly reaction. Elute in 30 uL molecular grade H2O.
- Dilute competent cell stock
- Dilute the 40 uL competent cell aliquot to 160 uL by adding 120 uL ice-cold water, then split the culture into four tubes (40 uL each). It is often easiest to work with 8- or 12-well strip tubes with the aluminum blocks to keep the cells cold and accessible. Add 5-10 uL source DNA from the PCR purification.
- Transformation
- Transform each diluted cell aliquot in the electroporator. Resuspend each cell aliquot in 250 uL LB and incubate at an appropriate temperature for the cell strain (often 37 C). Spread all 250 uL on an appropriate selective LB agar plate using sterile glass beads.
Misc
- settings for E. Coli recommended by BioRad customer support:
- brown-caped 1 mm gap tubes:
- 1.8 kV, 200 Ohms, 25 uF.
- green-capped 2 mm cuvette: (in case you don't have brown cuvettes ready)
- 2.5 to 3.0 KV, 200 Ohms, 25 uF
- brown-caped 1 mm gap tubes:
- Alternately, add ~950 uL ice cold SOC media to cuvette after shocking
- Optional: after shocking, chill sample on ice for 2 mins to permit the cells to recover.
- Optional: plate transformation onto prewarmed LB-agar plate supplemented with appropriate antibiotic. I generally plate 200μL but appropriate plating volume depends on efficiency of the transformation.
- Leave remaining SOC-cell mixture on the benchtop overnight.
- If you don't have any transformants, plate the rest of the transformation in the morning.
- Note: you will usually get plenty of colonies by using TB (not SOC media, not chilling cuvettes, not icing sample after applying voltage, not pre-warming plates.
- When choosing the # of vials and cuvettes, you can include 2 vials for two negative controls (one with no DNA added, and another with only cut vector added).
- Cuvette types: (see pic on right)

Resources
- Electroporation Protocols for Microorganisms, 1995. (eBook available for UW)
- background with references
- BioRad Movie about electroporation. Though it is made for a different machine than we have, the principles are applicable.
bio.net "essay" on electroporation parameters
- (source )
- Note: this protocol is not specific to E. coli. In fact, it appears to be written by an expert eukaryotic cell scientist.
- Interesting quotes:
- "cells survive better when treated with pulses of low amplitude/long duration than those of high amplitude/short duration."
- "using lower volumes gives longer pulses."
- "Another way of increasing pulse duration is to use electroporation medium with low ionic strength."
- "The probability for the DNA to get to the pore increases with increasing both cells and DNA concentration."
- "At some point, however, increasing DNA concentration becomes inhibitory. Two reasons for this: i) increasing concentration of "crap" contaminants that come with plasmid preps, ii) DNA is cytotoxic during electroporation because it, being a very large molecule forced through a very small electropore, enlarges pores sometimes to the point that they become irreversible."
- "So the best you can do is to ensure that DNA is as clean as possible."
- "At some point, however, increasing DNA concentration becomes inhibitory. Two reasons for this: i) increasing concentration of "crap" contaminants that come with plasmid preps, ii) DNA is cytotoxic during electroporation because it, being a very large molecule forced through a very small electropore, enlarges pores sometimes to the point that they become irreversible."
- "While no postpulse incubation is necessary, ~ 5 min after pulse does not affect survival significantly.
- In my experience, cells should not be diluted right away into warm culture medium. Use RT medium and let pores reseal for ~ 20 min before placing cells in 37C CO2 incubator."
- "By far the trickiest issue is electroporation medium.
- "Effect on the level of electricity is simple: in low salt the pulse is much longer then in high salt. All other effects are on the level of cell survival and biological/transcritptional responses downstream."
- "cells survive better when treated with pulses of low amplitude/long duration than those of high amplitude/short duration."
Electroporation Protocols for Microorganisms
Electroporation Protocols for Microorganisms, 1995. (eBook available for UW)
- "Try to avoid trapping air bubbles in the cell mixture once in the electroporation cuvet. The presence of air bubbles can lower the transformation efficiency. This results in a lower than expected time constant."
- "(Some growth medias include) addition of glycine or threonine to the growth medium as a cell-wall-weakening agent (2-4). Although effective for many species and strains, the susceptibility of each strain to glycine needs to be determined independently, since high glycine concentrations can cause cell lysis during the electric pulse (2)"
- (2) = Dunny, G. M., Lee, L. N., and LeBlanc, D. J. (1991) Improved electroporation and cloning vector system for Gram-positive bacteria. Appl. Environ. Microbiol. 57, 1194-1201. "Group A streptococci and B. anthracis transformed without glycine and actually lysed during electroporation when grown in glycine."
