BISC219/F13:Gene Mapping

An Investigation in Classical (Forward) Genetics: Linkage Analysis, Mapping the Mutation, Complementation Testing, DNA Sequencing Analysis, Bio-Informatics
In Project 2, you will progress through the normal sequence of events in forward (classical) genetics. Forward genetics investigations work to tie a gene to its function. This type of work generally starts with finding a worm with an aberrant phenotype (mutant) that is likely to be caused by a defect in a protein encoded by a mutated gene. In our investigation we will work to first locate a mutated gene in such a mutant in order to characterize the exact change in the gene (mutation) responsible for the aberrant phenotype. Working with abnormal genes/proteins facilitates understanding of the importance of the gene, gene product or functionally significant gene region in C. elegans and in other species. When we are able to make these structure/function connections by studying mutant worms and identifying the genes responsible for the defects, the main goal is not so much to understand defective gene function in worms, but rather, to be able to extrapolate the function of normal genes by seeing what their gene products are unable to do when altered. We are interested in worm genes because the genome of most eukaryotes astonishing similar. Many worm genes have homologs/orthologs in other eukaryotic species, including Homo sapiens.
To do this involved forward genetics study we start with an interesting phenotypically aberrant worm. One of our main goals is to pinpoint the location of the mutation on a specific gene. In order to do this we first must find out through linkage analysis the relative location of the gene associated with the mutation: which chromosome is it on? We will be able to find out if the aberrant phenotype is caused by a mutated sex-linked or autosomal gene and, if autosomal, on which autosome the defective gene is located. Our eventual goal is to pinpoint the exact location of this mutation in a gene on a chromosome (to map the mutation). We will use complementation analysis to find out, possibly, the identity of our gene of interest if the gene has been previously characterized. If not, our gene might, possibly, be a newly discovered one. To conclude our function/structure analysis, we hope to identify the exact change in the bases of the gene that causes its product to function abnormally. We will find the exact mutation through DNA sequencing of the mutated gene and by comparing it to the wild type sequence. From this sequencing and bioinformatics work, we may be able to extract new information about functionally significant regions of the gene and its product. This structure/function relationship may be significant in a broader context than in just our nematode species. We will look for homologs/orthologs in other species.
To begin, you will first examine a plate of mutant worms and determine how they are different from wild type. You will then pick your mutants to a separate plate, confirm that they are true breeding (meaning that the worm has two copies of the defective gene), and begin our forward genetic analysis. The first step in identifying a new gene associated with an interesting mutant phenotype, is usually a long, tedious process that requires applying a mutagen (UV or some mutagenic chemical like EMS) to wild type worms and then looking through thousands of normal worms to find a good candidate mutant. To make this study easier for you, mutants have been found and isolated for you and you just have to move some to a new plate to ensure they are true breeding.
Once you have recovered your mutant and confirmed its phenotype as true breeding (by examining its progeny), you will next perform linkage testing: determining on which chromosome (linkage group) your mutation is located. This task is a prerequisite to mapping. You must find out the relative location of the gene responsible for the mutation (find the chromosome it is on) before you can determine the exact location of the mutation on a gene. Linkage Analysis is accomplished by determining the segregation behavior of your unmapped mutation relative to standard reference markers (e.g., mutations whose location is already known). Recall that unlinked genes (on different chromosomes) will segregate independently (your basic dihybrid inheritance as first observed by Gregor Mendel) whereas linked genes (on the same chromosome) will not.
In practice, linkage tests are performed using the following steps (where "d" (dpy) represents your recessive mutant tested with a set of reference markers "u" (unc)). The markers d and u must be visibly distinguishable. Since homozygous mutant males usually will not mate, the desired double heterozygote is constructed by mating males heterozygous for your dpy mutation but wild type for all other genes including the reference mutation (d/+;+/+) with hermaphrodites homozygous for the reference mutation unc (+/+; u/u). The genotypes of the F1 hybrids will be (+/d;u/+) and (+/+;u/+). We are only interested in the double heterozygote (+/d;u/+). The F1 hybrids containing only u are not useful. To select the (+/d;u/+) heterozygotes, we let 4 to 5 individual F1's self fertilize on their own individual plates (one on each plate). We score the progeny of the F1 individuals (the F2) for linkage. Only F1 worms which produce d/d homozygotes are scored, since those are the (+/d;u/+) parents. The d/d homozygotes should be found on 50% of the plates.
F2 progeny of each class are counted in the (+/d;u/+) plates: wild-type (+/+;+/+); d (d/d;+/+); u (+/+;u/u) and du double (d/d;u/u). If assortment is independent, progeny will be:
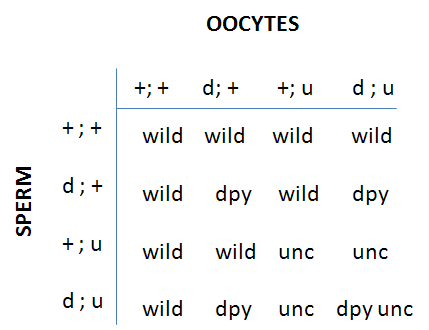
Or 9/16 wild; 3/16 d, 3/16 u; 1/16 du (that is our good old friend the 9:3:3:1 ratio)
On the other hand, if the markers are closely linked, double homozygotes (d u/d u) would occur only through two recombination events. Such an event might occur in both the sperm or the occyte and those recombinant gametes must come together. If the probability of a recombination event is p, and if the event produces wild-type and double mutant recombinant chromosomes, then the probability of getting the double mutant chromosome in an individual gamete is p/2. The chance of an individual gamete which is a (d u) recombinant combining with another (d u) recombinant is (p/2) x (p/2). If the map distance between 2 mutations is 10%, then the probability (P) of recombination occurring is p = 0.1. p/2 is 0.05 and (p/2) x (p/2) is 0.0025. Consequently only about 2 - 3 worms in a thousand will be double mutants if the genes were 10MU apart. That is significantly lower than the 63/1000 one would expect if the genes were not linked (1/16 of the progeny). Therefore, the test for linkage is usually the virtual if not complete absence of the double mutant class (d u/d u). You will use this first assesment to determine which chromosome the dpy mutation is on (the chromosomal location of each of the unc mutations is known).
Once you determined on which chromosome your dpy and unc are (for successful mapping they must be on the same chromosome), we will perform two factor mapping, which locates the gene of interest to a position on a chromosome relative to a known gene. In our investigation, to locate our gene on the chromosome we identified through linkage analysis, we first must construct a double mutant (du/du) that has phenotypically distinguishable mutations on a known gene and on our gene of interest. The appearance of a double mutant in the F2 requires the union of 2 recombinant gametes -- a very rare event in the cross in linkage analysis that identified the chromosome. It is unlikely that we will find a double mutant among the F2 progeny when the mutations are linked. The probability of a recombination event having occurred on one of the two homologues is much better; that is, there will be many more progeny that are genotypically du/d+ or du/+u than du/du. These Dpy or Unc mutant offspring exhibit one mutation's phenotype and segregate double mutants (du/du) as one quarter of its self-fertilization progeny. To find such a required double mutant for mapping, we choose 5 individuals of the Unc phenotype from the F2 linkage testing plate that shows no double mutants and allow those Unc hermaphrodites to self fertilize. We look for the segregation of the du/du double mutant among their progeny to get one parent for our mapping test cross.
To get the other parent for mapping, one crosses a homozygous double mutant hermaphrodite (du/du) with wild type males. The heterozygous F1 males that result are the required male parent. They will be mated to double homozygous recessive Dpy Unc hermaphrodites in a test cross and the number of F2 individuals of each different phenotype are then counted. We expect four phenotypes to be observed: wild type, Dpy, Unc, and Dpy Unc. The map distance between the unknown gene of interest and the known marker gene can be calculated any number of ways. You will determine map distances using the formula: RF (recombinant frequency) = the number of single mutants (dpy and unc single mutants totals) divided by the total number of worms counted * 100 (to obtain it in % recombinants and thus in map units). You will then go to WormBase and see what genes are found at that position up and downstream from the known marker gene on the chromosome you identified in linkage testing. Is there a previously identified gene or ORF at these positions. What is known about the function of the genes at these positions? Have you discovered a new gene or new function for a gene?
You will use complementation analysis to, perhaps, discover the identity of a previously characterized gene or to help you decide if your mapped locus is upstream or downstream from the reference gene. You may be able to discover the name of your gene through complementation analysis before you locate it through mapping. We are doing complementation testing because It is not unusual to have series of mutations that confer similar phenotypes to also map to a identical or similar location on a chromosome. Complementation testing can determine if two mutations are allelic (that is, in the same gene) or non-allelic (in different genes but both causing the same phenotype). This is done by crossing a mutant with a series of reference strains. In our case, we will use several different reference mutant strains. All have a Dpy phenotype, but in each strain the gene responsible for the dumpy defect has been located to a different known region of a chromosome.
If the mutations (your unknown's and that of a reference strain) are allelic (in the same gene but on different sister chromatids) there should be no complementation, meaning that the mutations can not "rescue" each other because you do not have a normal copy of the gene causing the mutant phenotype; thus, you observe the Dpy phenotype when the two strains combine.
If the mutations are non-allelic (in different genes) there should be complementation, meaning they "rescue" each other: because there is one normal copy of each defective recessive gene, the product of the two parental homozygous dpy mutants can show a wild type phenotype.
Complementation analysis can be evaluated this way only when you are working with recessive mutations.
The specifics of strain construction for complementation analysis vary depending on the experimental organism; however, the basic strategy in all cases is to construct a double heterozygote organism and then to examine the phenotype of this organism. Remember that a wild-type phenotype indicates that the two mutations complement (rescue) one another and are, therefore, in different genes. Conversely, a mutant phenotype suggests the mutations are allelic to one another (that is, they fail to complement).
We will construct different double heterozygotes containing your dumpy mutation of unknown location (dpy-u) and dumpy mutations of known location (dpy-k) on the same chromosome as your unknown dpy mutation as follows:
First you will obtain heterozygotes for your dpy mutation of unknown location through
Cross #1: unknown Dpy hermaphodites (dpy-u/dpy-u) x N2 males (+/+) yields dpy-u/+ progeny
Then you will use those heterozygous males resulting from Cross #1 in a subsequent cross:

Cross #2: dpy-u/+ males x a reference set of known Dpy hermaphrodites (dpy-k/dpy-k). These reference worms must have a defective gene causing the phenotype on the chromosome you identified by linkage analysis.
The main consideration is whether or not there are worms of Dpy phenotype present in the progeny of cross #2. If so, this is your "proof" of failure to complement meaning that the reference mutation and the mutation of interest are allelic (in the same gene). Since the reference mutation is in a known gene, you now know the name of your unknown gene of interest and that it is a previously characterized Dpy gene. Or if you don't have Dpy progeny you know that the mutations were complementary, eg. the the wild type phenotype was rescued by having mutations in different genes. In this case, you haven't learned what we wanted to know except that it is more likely that your unknown is a uncharacterized mutation.
DNA sequencing of the mutated gene or open reading frame (ORF) that we located by mapping (and possibly identified by complementation analysis) and bio-informatics will be used to identify and characterize the exact change in the DNA code.
Links to Labs & Project Info
Worm Info
Lab 1: Worm Boot Camp & Sex-Linked or Autosomal Start Project 1
Lab 2: Sex-Linked or Autosomal Finale Project 1/ Mutant Hunt Start Project 2
Background Project 2: Classical Forward Genetics
Lab 3: Linkage Test Part 1
Lab 4: Linkage Test Part 2, Mapping and Complementation
Lab 5: Finish Complementation; Mapping Continued
Background Information on Project 3: Investigating Gene Regulation Using RNAi
Lab 6: DNA sequence analysis; Mapping Continued/Obtain CL2070 worms & propagate
Lab 7: Complete Mapping & Project 2: Score/ Examination of Cl2070 hsp-16.2::GFP worms
Media Recipes
Lab 8: Creating the feeding strain of bacteria for RNAi
Lab 9: Induction of feeding strain to produce dsRNA
Lab 10: Phenotypic analysis of treated vs untreated worms
Lab 11: Optional Writing Conferences
