BISC209/S13: Lab2

LAB 2: Enumerating Microbial Members & Isolating Bacterial Members of a Soil Community
Today you will complete your first investigative goal in our semester long project: assessing abundance of a soil microbial community by enumerating its microbes by two methods, one culture dependent and one culture independent. Today you will also continue acquiring and practicing skills used by working microbiologists. In addition, you will continue experiments along our other lines of investigation, specifically, our culture-dependent approach to isolating a few bacteria of interest from your soil community in order to provide examples of richness and community co-operative and competitive behavior.You will continue the process of selection and isolation to pure culture of a few of those bacteria for the next several weeks. Next week we will begin our experiments on the whole community of soil microbes: a community level, culture dependent assessment of a few aspects of richness and cooperative and competitive behavior.
Finishing the Standard Plate Count of Culturable Soil Microbial Community
Activity: Last week you started a standard plate count of the culturable microbes in your soil sample on dilute nutrient agar. Today you will complete that plate count to get one kind of enumeration of the microorganisms in your soil community. Each pair will find the dilution of the serial dilution plates that contains 30-300 colonies. If it is clear that a culture plate has well over 300 colonies or under 30, designate it as "invalid" in your lab notebook and on the bottom of the plate. Count all the surface and subsurface colonies on the replicate valid plates. The colonies can be more easily counted by using a Quebec Colony Counter which allows proper illumination, a grid overlay and by slight magnification of the plate surface. (There are two colony counters in the lab.)
Calculating the number of colony forming units (CFU) (culturable bacteria) per gram of soil
For each plate counted (2 per pair, 4 per site), if you divide the number of colonies counted by the amount of inoculum plated times the dilution factor of that plate, you will obtain the number of cultivatable bacteria per gram of soil.
number CFU/(dilution plated*dilution factor) = number of CFU/gram
For example, if you counted 150 colonies on the 10-6 plate the calculation is:
150/(0.1ml inoculum*1X10-6dilution)= 150X107 which in scientific notation is written as 1.5X109 CFU/gram
Record the number of CFU/ gram of wet soil in your lab notebook and enter your data on the course spreadsheet on the instructor's computer at the front of the lab. Calculate the average and standard deviation for the plates from your sample site. Add your average and standard deviation data to the chalkboard for comparison with the other soil sampling sites. Note that this number is the average colony forming units/ gram WET soil for your sample site.
Soil bacteria are usually not recorded as number of colony forming units (CFU) in 1 gram of WET soil but instead as per gram of DRY weight. Therefore, you will need to figure out your counts as DRY weight. Please weigh each of the three 1 gram samples that you left last week for oven drying. Average the new dry weights. The weights should be considerably less than 1 gram. Save the 3 dried soil samples after you weigh them, you will need them again today.
Determine the % change in soil weight by subtracting the average dry weight from 1 g wet weight divided by the wet weight, then times 100.
[(wet weight - dry weight)/wet weight] * 100 = % change
Use this % change to convert the CFU per gram wet soil to CFU per gram dry soil. For example: If your dry weight soil averages 0.75 grams, then (1g-0.75g)/1g * (100) = 25% change. The number of microbes should be 25% higher than the number calculated above/gram of wet weight. Calculate 25% (or whatever your conversion factor is) of your CFUs and add that number to the CFUs/ gram of wet weight soil.
Record the number of CFU/ gram DRY weight soil in your lab notebook and add this information to the chalk board. If you are unsure of the accuracy of your calculations of CFUs per gram of wet soil weight and per gram of dry weight, check with your instructor. You will need accurate counts to set up community profiling analyses in LAB3.
Things to Consider:
Why did we perform so many dilutions when we set up our plate count in Lab 1?
Why should you have only one dilution with 30-300 colonies?
Why did we make up to 4 replicates per site and why do we list each count separately in the Google doc?
Seal the edges of the dilute nutrient agar plate that your group counted with parafilm and store the plate in the cold room. Don't discard any plates yet.
Enumeration of Community Soil Microorganisms by Direct Count of Microbial Genomes Stained & Viewed by Fluorescence Microscopy
You can directly count a random sample of microbes from the soil extract that you prepared last week and extrapolate the number per gram of soil. To make the microbes easier to count, your lab instructors stained the nucleic acids of your soil community microbes (not just the bacteria) with a fluorescent 4'-6-Diamidino-2-phenylindole (DAPI) DNA stain. All the microbes in a 1ml aliquot of the 1% soil extract that you prepared last week were transferred in a Poisson distribution to a small piece of filter paper. She viewed the fluorescent cells as discreet bright "spots" in several different areas of the filter paper distribution using fluorescence microscropy. These areas were photographed. These photomicrographs will be made available to you and your partners today. You will each count the discreet "spots" as individual microorganisms (one fluorescent genome/cell) from one area and perform the calculations described below to assess the total microbial concentration in this culture-independent enumeration of your soil community's microorganisms. Compare this number to the calculation of CFUs/gram of wet soil (a culture-dependent assessment) that you will also obtain today. Compare the two counts that, in theory, should be the same since we are using two methods to figure out the same thing: how many microbes per gram comprise your soil community. The answer will provide evidence for one of our investigative goals: abundance or microorganisms in your soil community. What does it mean if your two experimental methods don't give the same answer?
DIRECT COUNT OF MICROBIAL CELLS USING DAPI DNA: PROTOCOL
NOTE: The staining and imaging were performed by your lab instructor last week according to the protocol described below (provided here for reference only). YOU WILL NOT PERFORM THIS IN LAB!
DAPI aqueous solution Stock conc. is 1mg/ml : 4'-6-Diamidino-2-phenylindole (DAPI) is known to form fluorescent complexes with natural double-stranded DNA.
- If the samples will not be processed for a while: You need a final concentration of 4% parformaldahyde solution that will act as a preservative for the microbial cells in your soil extract samples. Ask your instructor about this step.
- Add 5μL of 1mg/ml DAPI stock (Thermo scientific prod. # 62248) stain to 1mL of a fresh 1% diluted soil extract so that the effective concentration of DAPI is 5 μg/mL (1μg/mL may work as well).
- Incubate at 4°C for 20 min in the dark.
- Set up a vacuum flask and filter apparatus (125 ml side-arm flask, Borosilicate base and fritted glass filter support with rubber stopper).
- Carefully place a 0.2μM glass fiber filter (Isopore membrane GTTP02500) onto the fritted glass filter support.
- Add the borosilicate glass funnel onto the base and clamp the two sections together using an anodized aluminum spring clamp.
- Rinse the filter once with 1 mL sterile deionized water using a vacuum pump to provide 178mm Hg of force. Wait until all the water is removed.
- Turn off the vacuum, gently break the vacuum by loosening the rubber stopper, re-tighten, and carefully transfer the 1mL of DAPI stained extract (made in step 1) onto the 0.2μM glass fiber filter.
- Turn the vacuum on and wait until all the solution is filtered.
- Rinse with 1 ml sterile deionized water to remove any background DAPI.
- Dissassemble the funnel, gently breaking the vacuum by loosening the rubber stopper.
- Use forceps to carefully remove the filter and place it on a microscope slide.
- Add 1 drop of Fluoro-Gel with Tris buffer (Electron Microscopy Sciences Cat #17985)and a coverslip.
- Use the Nikon Eclipse 80i fluorescent microcope in L318C to view and photograph the DNA at 1000x magnification.
- Use the Nikon NIS-Elements Imaging software, to photograph a field of view in white light (this allows you to see relative size and shape of the organisms in the sample, remember some will be eukaryotes), save this image, then do the same using the UV light at 350nm excitation wavelength (filter #1) so you can visualize the fluorescently labled DNA.
- Save all images to a 209-2012 file folder.
- Count the unique spots of blue fluorescence; each indicates a soil microbe's genetic material (chromosome or nucleus). It is assumed that the bacteria are arranged in a Poisson distribution. For the most accurate counts, 20 fields or 400 bacteria should be enumerated to determine the number of bacteria per ml.
COUNTING & CALCULATIONS:
The area of each field of view at 1000X using the Fluorescent scope is 10487 μmeters2. The area is determined for the microscope used, in our case, a NIKON 80i fluorescent microscope. The diameter of the filterable section of the borosilicate apparatus is 17 mm (8500 μmeter radius). Therefore, the area is 2.28 X 108μmeter2. Multiply the number of microorganisms counted on the photomicrograph by a factor of 2.17X104 (2.28 X 108μm2 divided by 1.0487 x 104μmeters2) to determine the number of organisms found in 1mL of filtrate of extract. Then correct for the 1:100 dilution factor of filtrate which is the number of organisms in 1 gram of wet soil. Convert this to the number of organisms in the community in 1 gram of dry weight soil.
Post your mean (average of the areas of the filter counted) number of microbes/gm of soil to the spread sheet on the instructor's computer and on the board (all calculations must also be in your lab notebook!) from both the culture-dependent and culture-independent enumerations. Report them as the estimated number of microorganisms in 1gm of wet weight soil and 1gm of dry weight soil. Record these numbers in your lab notebook in scientific notation and as numbers (with an amazing number of zeros). Consider the relative insignificance of one gram of anything and the enormity of the number of microbes that thrive in a gram of soil! How is it possible that each of the microbial members of such a soil community can find a niche, obtain all the nutrients needed to grow and reproduce, and contribute to overall health of the soil and to the community of microorganisms?
Now that we have some sense of the abundance of microbes in a soil community we can move on to investigating the richness (diversity) and the co-operative and competitive behaviors that maintain it.
Adapted from Schallenberg, M., Jacob, F. and Joseph B. R. (1989) Solutions to Problems in Enumerating Sediment Bacteria by direct counts. Applied & Environmental Microbiology. p. 1214-1219.
RICHNESS OF A MICROBIAL SOIL COMMUNITY & METABOLIC DIVERSITY:
Isolation & Characterization of Cultured Bacteria from a Soil Habitat
Using General Purpose & Enrichment/Selective Media for Isolation and Identification of Soil Bacteria in a Mixed Population
Please watch the YouTube video on streaking for isolation and pay attention to your instructor's demonstration: http://www.youtube.com/watch?v=eyoW18Fzb3o
Directions for Streaking for Isolation are found in the Protocols section of this wiki.
Streaking for Isolation from a Dried Soil Extract:
Each team of students will make a soil extract from one gram of their oven dried soil sample.
Aseptic transfer and streaking for isolation are crucially important skills in microbiology. When you are ready to streak out your first plate (after you have read the directions carefully, asked questions to clarify any confusion, and practiced your best technique on an empty plate or piece of paper), call your instructor over and ask her to watch you streak out your first plate. She will give you pointers to enhance your success at repeating this technique on a second plate. Your goal is to have well-separated single colonies when you evaluate your streaked plates next week. Label the plates with your initials, date, lab section, soil sample identifier information, and medium name. Incubate one plate at room temp and one plate at 30°C in the racks assigned to your group or Lab section.
Enrichment & Isolation of endospore-forming bacteria from oven dried soil extract on Glycerol Yeast Extract (GYEA) Medium: 0.5% (v/v) Glycerol, 0.2% Yeast Extract, 0.1%K2HPO4(Dipotassium phosphate), 1.5% Agar
Endospore forming genera include Bacillus and Streptomyces as well as other members of the Actinomycetes family
Oven drying will have killed most of the vegetative non-spore forming microbes. GYEA medium is particularly selective for the Streptomyces group of endospore formers because of the osmotic pressure obtained from the high concentration of glycerol. Few bacteria can thrive in GYEA which favors the growth of members of the Streptomyces group of Gram positive bacteria.
In pairs set up enrichment/selection cultures for spore-forming bacteria on two plates of glycerol yeast extract agar (GYEA) following the directions below (also found in the Enrichment Media for the Isolation of Soil Bacteria in a Mixed Population: Finding Spore Forming Bacteria in the Protocols: Culture Media section of this wiki).
Isolation of Spore-formers by isolation streak:
- You will use a new sterile cotton swab dipped into the oven-dried-soil extract to Swab section 1 of a pre-labeled plate of glycerol yeast medium (GYEA).
- Wait one minute to allow the inoculum in section 1 to absorb into the agar before you switch to your inoculating loop to streak from section 1 into section 2, following the steps for Streaking for Isolation in the Protocols section of this wiki.
- Flame your loop and isolation streak section 2.
- Flame and Repeat to streak sections 3 and 4.
- Invert, and incubate the plate at RT.
- Check your plate for colonies daily and record your descriptions of the texture and shape of the colonies that appear. If any colonies arise that look tough and leathery or appear as "little, powdered-sugar volcanos", use the tip of a sterile toothpick to pick up a small but visible amount of growth (being careful not to touch anything but the tip of the colony) and "spread" the bacterial growth onto section 1 of a new glycerol yeast plate. (Inoculate one colony/plate.) Use your loop to isolation streak sections 2-4. Don't forget to flame your loop between sections!
- We will select colonies of other potential spore-forming bacteria in lab 3.
Isolation of Spore-formers by Spread Technique:
- Transfer 100 µL of soil extract onto a pre-labeled plate of glycerol yeast medium (GYEA).
- Using a sterile disposable spreader cover the surface of the plate with the inoculum.
- Wait one minute to allow the inoculum to absorb into the agar.
- Invert, and incubate the plate at RT.
- Check your plate for colonies daily and record your descriptions of the texture and shape of the colonies that appear. If any colonies arise that look tough and leathery or appear as "little, powdered-sugar volcanos", use the tip of a sterile toothpick to pick up a small but visible amount of growth (being careful not to touch anything but the tip of the colony) and "spread" the bacterial growth onto section 1 of a new glycerol yeast plate. (Inoculate one colony/plate.) Use your loop to isolation streak sections 2-4. Don't forget to flame your loop between sections!
- We will select colonies of other potential spore-forming bacteria in lab 3.
Over the next few weeks you will continue to sub-culture onto new plates, using your best isolation streak technique. Your goal is to continue to streak out ONE CFU until you are sure that all the bacterial growth in a colony comes from a single mother cell (pure culture). In subsequent labs you will make a bacterial smear and do a Gram stain of these genetically identical bacteria and you will perform other tests from freshly pure cultures to explore the physical and metabolic characteristics of this isolate.
Streptomycetes and other Actinomyces bacteria often form tough leathery colonies, so transfer of these bacteria to new media to start a sub-culture is sometimes difficult. The powdery area may be spores. Take a sample from this area, if possible. In any case, try to "break off" a piece of the colony with your sterile loop or with a sterile toothpick and transfer that whole piece of a colony onto zone one of the new plate. Then use your loop for streaking out the other zones. The tiny spores on the surface of the colony are likely to transfer to the next plate or tube when you work with it. (That's a good thing this time.)
Your team will obtain additional Streaking for Isolation practice by following the directions for SECONDARY ENRICHMENT and ISOLATION using the broth culture and the spread plates that you inoculated with your soil extract last week. This process will continue your attempt to find a diverse group of nitrogen cycling and other interesting bacteria in your soil community. You and your teammates should divide up the work described below and in the Enrichment Media for the Isolation of Soil Bacteria in a Mixed Population.
The directions for Aseptic transfer: Broth to Broth and Broth to Plate are found in the Protocols section of the wiki.
Finding Nitrogen Cycling Bacteria
Nitrogen Fixers: Using Azotobacter Nitrogen Free Medium:
0.08%K2HPO4; 0.02%KH2PO4, 0.02% MgSO4 0.01% CaSO4; 0.0015% FeSO4/7H2O; 0.00025% g MoO3; 0.5% sucrose (to select for nitrogen fixers).
Note that the ingredients of this medium are not extracts of undefined sources such as beef digest but rather defined chemicals. The medium contains no nitrogen and only one carbon source-- one could use any sugar, but we will use sucrose. Microorganisms that can't use sucrose for all of their carbon needs and that can't fix nitrogen from the atmosphere to synthesize proteins and other required nitrogen containing compounds will not grow on this medium. Those that can, such as the Azotobacter group will be selected.
- Secondary enrichment:
-
Each pair should take a loopful of the slime from the pellicle (media surface) or from the side of the flask and place it into 1 ml of sterile water in a small tube. Cap the small tube. Vortex the tube to disperse the sample. (Vortexing in this way helps break up the other microbes that will be embedded in the slimy material. The other microbes are taking advantage of the by-products of Nitrogen compounds excreted by the N2 fixers. )
OR use a sterile swab to remove some of the slime and a little liquid from the flask of Azoto medium and inoculate the first zone of an isolation streak plate directly.
- Each Pair should streak onto two plates of Azotobacteria agar medium suspension, using your best isolation streak technique. (Follow the protocol in Streaking for Isolation ).
- Incubate one streak plate at room temp and the other at 30 °C. Use the shelf in the 30°C walk in chamber that is designated for your 209 lab section.
Isolation:
Watch for the appearance of isolated, slimy colonies on either plate.
Continue to streak to make sub-cultures onto fresh Azotobacter medium or on NA until you are sure that you have a pure isolate. Note that your bacteria will probably grow better on NA and it is fine to switch to nutrient agar after the initial selection.
Characterization:
In a later lab you will make a bacterial smear and do a Gram stain and other tests to explore the physical and metabolic characteristics of your nitrogen fixing bacteria.
Finding Nitrogen Cycling Bacteria: Bacteria that can Extract Nitrogen from Ammonium Compounds
Ammonium & Citrate Users: Using Simmons Citrate Medium:
0.02% MgSO4(Magnesium Sulfate), 0.1% NH4H2PO4(monoammonium phosphate), 0.1% K2HPO4(dipotassium phosphate), 0.2% C6H5Na3O7(sodium citrate), 0.5% NaCl(sodium chloride), 2.5% agar, 0.008% C27H27Br2O5SNa(bromothymol blue) at pH6.9
Simmons Citrate Medium selects for microorganisms that can utilize citrate as their sole source of carbon in a medium containing inorganic ammonium salts as its only source of nitrogen. In 1923 an investigator named Koser invented a broth medium in which ammonium phosphate supplied the only source of nitrogen and each organic acid was added individually, allowing the introduction of carbon utilization as a diagnostic aid. Later, an investigator named Simmons converted Koser’s liquid medium to a solid by the addition of agar and added an indicator system by incorporating bromthymol blue. The exact nature of the alkaline reaction produced by the organisms that are able to use citrate as their sole source of carbon is poorly understood. It appears that the alkaline reaction that gives the color change characteristic of a positive Simmons-Citrate test most likely occurs when excess CO2 is generated when citrate is cleaved to form oxaloacetate. This by
product is then decarboxylated to pyruvic acid and CO2. The excess CO2 combines with sodium and water to form sodium carbonate. Note that bacteria that utilize citrate in this way are able to extract nitrogen from ammonium phosphate in the medium, resulting in the production of ammonia which combines with water to form NH4OH. These reactions in combination produce an alkaline pH (greater than 7.6), resulting in a color change in the indicator from green to blue. Some members of the generaKlebsiellain the Entrobacteriacea family are Simmons Citrate positive.
Isolation of Ammonium & Citrate Users from Simmons Citrate Selective Medium:
- Count the CFU on a plate with 30-300 colonies and using a Sharpie circle and number a few of the different appearing ones on the bottom of the plate or give them code names (your initial and a number or a more creative coding scheme).
- Some of the colonies on this plate may floresce. In order to see this, you will need to take a marker, safety goggles, the hand-held UV light on the instructors' desk, and your SC plate culture to a completely dark location (such as the bathroom in the hall outside the lab). Put on the glasses, turn on the UV light and shut off the white lights. If you see any colonies that "light up"(B), try to mark them with a small dot or circle on the bottom side of the plastic plate.
A B
B 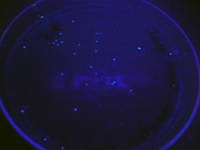
- Make observations in your lab notebook about the #, appearance, color, consistency or florescence of any colonies growing on your Simmons Citrate plates.
- Each teammate should try to isolate a unique colony from any of the Simmons Citrate plates (based on colony color, consistency, and/or florescence).
- Obtain new sterile Nutrient Agar plates or SC plates (one for each student in your group).
- Each student should use her flame sterilized and cooled loop and touch it to the center of the colony on Simmons Citrate that you are attempting to isolate. DO NOT touch anything on this plate other than the desired colony!
- Follow the directions found in Streaking for Isolation in the Protocols section of this wiki carefully. Transfer bacteria from this colony to the O zone of a new NA plate.
- Flame and cool your loop before going back into sequential, inoculated zones to transfer fewer and fewer bacteria as you streak each zone.
- Parafilm and save any plate that contains a colony you or your teammates are attempting to isolate. Discard unused cultures.
- Check the sub-cultures frequently and sub-culture over and over to new media until you are sure that your culture is pure. Note how fast your organism grows and if the initial colony appearance and/or fluorescence is maintained.
- Your goal is to end with well isolated single colonies that grow from a single bacterium (all genetically identical) on nutrient agar (NA). It will probably take several transfers over the next few weeks to take a CFU from this original selective medium plate and grow bacteria from it in pure culture on nutrient agar.


Finding and Isolating Other interesting Bacteria to Pure Culture:
NA: 0.3% Beef extract, 0.5% Peptone, 1.5% Agar; Deionized water to 1 L at pH 6.6- 7.0 at 25°C
NA+Starch: NA (above) plus added 2.5% starch
Dilute NA: NA diluted 1:10 (0.03% beef extract, 0.05% Peptone, 1.5% agar)
Nutrient Agar is a rich, general purpose medium. It is not used for enrichment or selection although not all microorganisms will grow on it. Those that won't are considered, with great variability, "fastidious". NA + starch is similarly non-selective and should grow the same microorganisms that grow on NA although you may find that the addition of starch encourages the growth of organisms that can use the starch effectively and/or observe interesting pigmentation to some colonies.
- Number on the bottom of the plate or use a code for the colonies each member of your group selects.
- Parafilm and save these plates in the refrigerator in your group's rack.
- Follow the directions carefully that are found in Streaking for Isolation in the Protocols section of this wiki. Transfer bacteria from one of your selected colonies to the O zone of a new NA plate.
- Flame and cool your loop before going back into sequential inoculated zones to transfer fewer and fewer bacteria as you streak each zone of the new plate.
- Your goal is to end with well isolated single colonies that grow from a single bacterium (all genetically identical) on nutrient agar (NA). It will probably take several transfers over the next few weeks to take a CFU from this original selective medium plate and grow bacteria from it in pure culture on nutrient agar.
The complete procedures for Enrichment and Isolation of Selected Bacteria are described in Enrichment Media for the Isolation of Soil Bacteria in a Mixed Population protocols).
For more information about culture characteristics of Azotobacter refer to on-line reference books such The Prokaryotes or Bergeys (link for access in the Resources section of the wiki. For an online example of images of colonies and stained cells of Azotobacter vinelandii [1] http://inst.bact.wisc.edu/inst/index.php?module=book&func=displayarticle&art_id=274
LAB NOTEBOOK CHECK
Your lab instructor will check your notebook today (not graded) and make suggestions for improvement.
CLEAN UP
1. All culture plates that you are finished with should be discarded in the big orange autoclave bag near the sink next to the instructor table. Ask your instructor whether or not to save stock cultures and plates with organisms that are provided.
2. Culture plates, stocks, etc. that you are not finished with should be labeled on a piece of your your team color tape. Place the labeled cultures in your lab section's designated lab area at room temperature (RT), the walk-in 30°C room, or the walk in cold room. Place any tubes in a labeled rack. If you have a stack of plates, wrap a piece of your team color tape around the whole stack and label it with your initials and the date.
3. Remove tape from all liquid cultures in glass tubes. Then place the glass tubes with caps in racks by the sink near the instructor's table. Do not discard the contents of the tubes.
4. Glass slides or disposable glass tubes can be discarded in the glass disposal box.
5. Make sure all contaminated, plastic, disposable, serologic pipets and used contaminated micropipet tips are in the small orange autoclave bag sitting in the plastic container on your bench.
6. If you used the microscope, clean the lenses of the microscope with lens paper, being very careful NOT to get oil residue on any of the objectives other than the oil immersion 100x objective. Move the lowest power objective into the locked viewing position, turn off the light source, wind the power cord, and cover the microscope with its dust cover before replacing the microscope in the cabinet.
7. If you used it, rinse your staining tray and leave it upside down on paper towels next to your sink.
8. Turn off the gas and remove the tube from the nozzle. Place your bunsen burner and tube in your large drawer.
9. Place all your equipment (loop, striker, sharpie, etc) including your microfuge rack, your micropipets and your micropipet tips in your small or large drawer.
10. Move your notebook and lab manual so that you can disinfect your bench thoroughly.
11. Take off your lab coat and store it in the blue cabinet with your microscope.
12. Wash your hands.
13. See you next time!
Assignment
Graded Assignment:
Write an explanation of the "Great Plate Count Anomaly" with citations and references. This assignment will become part of your final paper, useful in both the Introduction and Discussion. Remember that you will NOT use the data you collected this week on the number of microbes in your soil community. You will use that evidence in next week's assignment.
Consult the full directions for this assignment found at: Lab 2 Assignment: Assignment: Great Plate Anomaly.
Do before next lab:
1. Someone from your team must go to the Greenhouse and collect a new soil sample from your sampling site. On the day of lab next week, BEFORE lab begins, stop by the lab to pick up a plastic bag containing materials for collecting a new soil sample from your sampling site in the greenhouse. You will only need enough soil to fill half of a sterile, small 15ml, orange capped, conical tube. Make sure you get the sample for THE SAME sampling site as your original sample. Again avoid the top few millimeters of surface soil and wear gloves to avoid contaminating the sample with bacteria from your skin. You probably won't need to use the corer; a disinfected metal spatula or even a spoon should work fine.
2. Goal for Cultured Bacteria Isolation: Characterize a variety of spore forming, nitrogen cycling, or other interesting bacteria in a soil community
One goal in this project is for your group to end up with 16 unique bacteria (4 each) found in the soil community you sampled. Work with your partners so that you each choose different looking bacterial colonies from each medium and keep consulting with each other to ensure that your subset is likely to be unique.
Isolation of desired bacteria from mixed culture is challenging. Your best organizational skills are required. You will be expected to come in at times, on your own, to start, continue or complete this process of isolating to pure culture. We will make every attempt to make the media and reagents that you require available when you need them, but that availability requires advance planning on your part as well as on ours. Communicating your needs or desires well in advance of time of use, will reduce frustration and speed up the process. Remember that because this is an investigative, project based lab course, your instructors do not know the identity of the bacteria you are culturing from your soil habitat. The success of this project is in your hands. Early and continual updating of the plan you devise is crucial. You will not turn in your plan for a grade next week, but your instructor would like for you to create a master plan or flow chart with a preliminary time line and have it in your lab notebook for reference.
Links to Labs
Lab 1
Lab 2
Lab 3
Lab 4
Lab 5
Lab 6
Lab 7
Lab 8
Lab 9
Lab 10
Lab 11
