20.109(F14): Mod 1 Day 5 Examine candidate clones & tissue culture

Examine candidate clones
Introduction
Miniprep
Yesterday a few milliliters of LB+AMP broth were inoculated with some candidate colonies and the tubes were grown overnight at 37°C. Ampicillin was included in the broth to ensure that the cells would maintain the plasmid DNA through continued selection. Today the cells will have grown to high density and the plasmid DNA will have undergone many replications. All the copies of the plasmid DNA in your overnight culture should be identical (“clones” of one another), since the culture began with a single colony and that colony grew from a single transformed cell. Next, you must isolate the plasmid DNA from the candidates and determine if any of the plasmids contain the truncated EGFP gene.
To isolate the plasmid from the overnight cultures, you will perform what is commonly called a “mini-prep.” This term distinguishes the procedure from a “maxi-” or “large scale-prep” which involves a larger volume of cells and additional steps of purification. The overall goal of each “prep” is the same--to separate the plasmid DNA from the chromosomal DNA and cellular debris, allowing the plasmid DNA to be studied further.
In the traditional mini-prep protocol (based on this paper), which you will perform today, the media is removed from the cells by centrifugation. The cells are resuspended in “Solution I,” which contains Tris to buffer the cells and EDTA to bind divalent cations in the lipid bilayer, thereby weakening the cell envelope. A solution of sodium hydroxide and SDS is then added. The base denatures the cell’s DNA, both chromosomal and plasmid, while the detergent dissolves the cellular proteins and lipids. The pH of the solution is returned to neutral by the acetic acid and potassium acetate in “Solution III.” At neutral pH the SDS precipitates from solution, carrying with it the dissolved proteins and lipids. In addition, the DNA strands renature at neutral pH. The chromosomal DNA, which is much longer than the plasmid DNA, renatures as a tangle that gets trapped in the SDS precipitate. The plasmid DNA renatures normally and stays in solution, effectively separating plasmid DNA from the chromosomal DNA and the proteins and lipids of the cell.
Tissue Culture
In the past century, we have learned a tremendous amount by studying the behavior of mammalian cells maintained in the laboratory. Tissue culture was originally developed about 100 years ago as a method for learning about mammalian biology. The term tissue culture was coined because people were doing exactly that, extracting tissue and letting it live in a dish for a short time. Today, most tissue culture experiments are done using isolated cells rather than whole tissues. Much of what we know about cancer, heritable diseases, and the effects of the environment on human health has been derived from studies of cultured cells.

What types of cells do people study, and where do they come from? Cells that are isolated from tissue are called primary cells, because they come directly from an animal. It is very difficult to culture primary cells, largely because primary cells that are placed in culture divide only a limited number of times. This limitation on the lifespan of cultured primary cells, called the Hayflick limit, is a problem because it requires a researcher to constantly remove tissues from animals in order to complete a study. Cell isolation processes can be quite labour-intensive, and also can complicate data analysis due to inherent animal-to-animal variation. To get around the first of these problems, people have studied cells that are immortal, which means that they can divide indefinitely. (Some inherent cell-to-cell variation still exists in such cells.)
One familiar type of immortalized cell is the cancer cell. Tumor cells continuously divide, allowing cancer to invade tissues and proliferate. Cancer cells behave the same way in culture, and under the right conditions, cells can be taken from a tumor and divide indefinitely in culture. Another type of immortalized cell is the embryonic stem cell. Embryonic stem cells are derived from an early stage embryo, and these cells are completely undifferentiated and pluripotent, which means that under the right conditions, they can become any mammalian cell type. Mouse embryonic stem cells have become a valuable research tool, and it is this cell type that we will be using for our current experimental module.
The art of tissue culture lies in the ability to create conditions that are similar to what a cell would experience in an animal, namely 37°C and neutral pH. Blood nourishes the cells in an animal, and blood components are used to feed cells in culture. Serum, the cell-free (and clotting-factor free) component of blood, contains many of the factors necessary to support the growth of cells outside the animal. Consequently, serum is frequently added to tissue culture medium, although serum-free media exist and support some types of cultured cells.
Cultured mammalian cells must grow in a germ-free environment and researchers using tissue culture must be skilled in sterile technique. Germs double very quickly relative to mammalian cells. An average mammalian cell doubles about once per day whereas a bacterium is able to double every 20 minutes under optimal conditions. Consequently, if you put 100 mammalian cells and 1 bacteria together in a dish, within 24 hours you would have ~200 unhappy mammalian cells, and about 100 million happy bacteria! Needless to say, you would not find it very useful to continue to study the behavior of your mammalian cells under these conditions!
One major objective for this experimental module is for you to learn how to perform tissue culture. You will learn how to get mouse embryonic stem cells to grow in a dish and also how to prevent contaminants from getting into your cell cultures. All of this will form the foundation for the ultimate experiment, which is to detect DNA rearrangements in mammalian cells by the appearance of a fluorescent signal.
Protocol
Two notes:
- The tissue culture facility in the lab can accommodate no more than 12 students at a time. Half the class will begin by performing the tissue culture portion of today’s experiment (part 4) while the other half will begin in the main lab preparing their samples to load on an agarose gel (parts 1 through 3).
- Sometime before you leave today you should count or estimate the number of colonies that arose from your transformation reactions and post these values to the Talk page.
Part 1: Plasmid miniprep
One of the teaching faculty has set up four overnight LB + AMP cultures of bacteria for you to work with. Three of these cultures have colonies from the ligation reactions you have been preparing (if your reactions were unsuccessful then you have been provided with candidates from other reactions). One of these cultures has grown from a colony of bacteria with pCX-NNX. Plasmid from this culture will serve as a control for today’s digests and will provide a suitable comparison pattern on the agarose gel we will run for you this evening.
- Label your three candidate cultures 1, 2 or 3 and four eppendorf tubes (control, 1, 2, or 3). Vortex the bacteria then pour some into the appropriate eppendorf tube so that the tubes are almost full. If you are nervous about pouring the liquid, you can use your P1000 to pipet 750 μL into each eppendorf twice. Either way, the eppendorf should be quite full when you try to close the cap. You can wear gloves to keep the bacteria from splashing your skin or you can wash your hands after closing all the caps.
- Balance the tubes in the microfuge, and then spin them for one minute.
- Aspirate the supernatant, as shown, removing as few cells as possible.
Aspirate the supernatant, as shown, removing as few cells as possible - Be sure to change the yellow tip on the end of the Pasteur pipet between each sample
- Resuspend the cells in 100 μl of Solution I, changing tips between samples.
- Prepare Solution II by mixing 500 μl of 2% SDS with 500 μl of 0.4M NaOH in an eppendorf tube. Add 200 μl of Solution II to each sample and invert the tubes five or six times to mix. In some cases the samples may appear to "clear" but don't worry if you don't see a big change. Place the tubes on ice for at least 2 minutes.
- Add 150 μl of Solution III to each tube and immediately vortex each tube for 10 seconds with your vortex set at the highest setting. White clumps should appear in the solution after you vortex it. Place the tubes in the room temperature microfuge and spin them for 4 minutes.
- While the tubes are spinning, label another set of eppendorf tubes with the plasmid names and your team color.
- A white pellet should be visible when you remove your tubes from the microfuge. Use your P1000 to transfer 400 μL of each supernatant to the appropriate clean eppendorf tube. It's OK to leave some of the supernatant behind. Avoid transferring any of the white pellet.
- Add 1000 μl of room temperature 100% ethanol to each new tube. The tubes will be quite full. Close the caps and invert the tubes at least five times to thoroughly mix the contents.
- Microfuge the samples for 2 minutes. It is important to keep track of the orientation of your tubes in the microfuge this time since the pellets from this spin will be barely visible.
- Remove the supernatants using your P1000 or the aspirator, but be careful not to disturb the pellet of plasmid DNA that is at the bottom of the tube. Remove as much of the supernatant as possible but you do not need to remove every drop since you will be washing the pellet in the next step.
- Add 500 μl of 70% ethanol to each pellet. Spin the samples one minute, orienting the tubes in the microfuge so you will know where to find the pellet. Immediately remove the supernatant with your P1000, making sure to keep the tip on the side of the tube that doesn't have your pellet. Remove as much liquid as possible, using your P200 set to 100 μL, to remove the last few droplets.
- It's a good idea to check with the teaching faculty whether you have removed enough ethanol.
- To completely dry the pellets, place your rack in the hood with the caps open for a few minutes. When the pellets are completely dry, add 50 μl of sterile water to each sample and vortex each tube for 2 X 30 seconds to completely dissolve the pellets. After each vortexing step, the liquid can be brought back to the bottom of the tubes by spinning them in the microfuge for a few seconds.

Part 2: Measure DNA concentrations
Nucleic acids can be evaluated by UV spectrophotometry. Specifically, both RNA and DNA have an absorbance peak at 260 nm. Beer's law may be used to quantify the amount of DNA from this peak: Abs = ε l c, where Abs is the measured absorbance, l is the path length (1 cm for most specs), c is concentration, and ε is the extinction coefficient. For DNA, ε is 0.02 (μg/mL cm)-1, so 1 absorbance unit corresponds to 50 μg/mL of DNA. The absorbance at 280 nm gives some indication of DNA purity, as proteins have their absorbance peaks at that value (primarily due to the aromatic peptides tryptophan and tyrosine). An Abs260:Abs280 ratio of ~1.8:1 is indicative of good quality DNA.
- For each DNA sample, you will add 6 μL of miniprepped DNA to 594 μL of water. This ought to give you a measurement that's in a reliable range. You can prepare your DNA dilutions directly in cuvettes.
- Notice that today we are using cuvettes made of a special plastic that is transparent to UV light.
- To get the most accurate measurement, you should also prepare a blanking solution that contains everything except the DNA, i.e., 594 μL of water and 6 μL of EB elution buffer.
- At the spec, enter Nucleic Acid Analysis mode. Option 4 should be highlighted, and you can hit START to proceed.
- Begin with the cuvette containing blanking solution, and hit Blank on the spectrophotometer.
- Proceed to take an absorbance scan of each DNA sample. Record the 260 nm and 280 nm absorbance values in your notebook. The spec will calculate the purity ratio for you, but not the DNA concentration.
- What assumption are we making about the cuvettes when we put the blanking solution in a separate cuvette from the samples?
- Calculate the three DNA concentrations and purity ratios. Don't forget to convert from diluted to stock concentration!
- Note that our home-brew miniprep does not purify DNA from RNA, so your concentrations will be overestimates that reflect contributions from both nucleic acids. Based on these values, choose two of your three candidates to proceed with. (Proceed at random if all three are equally good!)
Part 3: Diagnostic digests
You will perform a diagnostic digest on three of the four plasmids you have prepared (the pCX-NNX parent and 2 of your 3 candidates) to see if any have the PCR insert. Use information from the lab manual, the NEB catalog and the plasmid maps you’ve drawn to choose the enzymes you’ll use. The following table may be helpful as you plan your work. As an example, XbaI is 20,000 U/mL; therefore one reaction will require 0.125 μL and a four-fold reaction cocktail will use 0.5 μL of the enzyme. Note: we always buy the "S" size and concentration of enzymes.
| Diagnostic Digest | Enzyme 1 only | Enzyme 2 only | uncut | |
|---|---|---|---|---|
| Plasmid DNA | 5 μl | 5 μl | 5 μl | 5 μl |
| 10X NEB buffer | 2.5 μl of buffer#_____ | 2.5 μl of buffer#_____ | 2.5 μl of buffer#_____ | 2.5 μl of buffer#_____ |
| Enzyme (2.5 U) | ____ μl of _____ | ____ μl of _____ | ||
| 2nd Enzyme (2.5 U) | ____ μl of _____ | _____ μl of _____ | ||
| H2O | For a total volume of 25 μl | |||
- Prepare a reaction cocktail for each of the above reactions (uncut, singly cut with enzyme 1, singly cut with enzyme 2 and doubly cut with enzyme 1 and enzyme 2) that includes water, buffer and enzyme. Prepare enough of each cocktail for 4 digests in order to have some excess. Leave the cocktails on ice.
- Aliquot 5 μl of plasmids (pCX-NNX and 2 of your three candidates) into a set of well-labeled eppendorf tubes. The labels should include the plasmid name, the enzymes to be added, and your team color.
- Add 20 μl of the appropriate cocktail to each tube. Flick the tubes to mix the contents, short spin down, and then incubate the mixtures at 37°C.
- Before leaving lab today, please add 2 μl of loading dye to each of the digests you have assembled. Store the digests and the DNA at –20°C and return the remainder of your bacterial cultures to one of the teaching faculty.
Part 4: Tissue Culture
Each of you will have a 60 mm dish of mouse embryonic stem (MES) cells that you will use to seed a six-well dish. You and your partner will seed the dishes at different concentrations so you should decide who will seed at 1:100 and who will seed at 1:400. We will have a short demonstration in TC before you begin your work.
- Prewarm all the required reagents in the water bath.
- Look at your cells as you remove them from the incubator. Look first at the color and clarity of the media. Fresh media is reddish-orange in color and if the media on your cells is yellow or cloudy, it could mean that the cells are overgrown, contaminated, or starved for O2. Next look at the cells on the inverted microscope. Note their shape and arrangement in the dish and how densely the cells cover the surface.
- Move the cells into the sterile hood, as well as the PBS, trypsin, and media that you will need. One of the greatest sources for TC contamination is moving materials in and out of the hood since this disturbs the air flow that maintains the sterile environment inside the hood. Anticipate what you will need during your experiment to avoid moving your arms in and out of the hood while your cells are inside.
- Aspirate the media from the cells.
- Wash the cells by adding 3 ml PBS with a 5 ml pipet. Tip the dish in all directions to rinse all the cells, and then aspirate the liquid out of the dish.
- Repeat the PBS wash, leaving the cells dry for the next step.
- To dislodge the cells from the dish, you will add trypsin, a proteolytic enzyme. Using a 2 ml pipet, add 0.5 ml of trypsin to the dish. For one minute precisely (use your timer), tip the dish in each direction to distribute the trypsin over the cells, and then aspirate the trypsin off the cells. Incubate the cells (“dry”) at 37° for 10 minutes, again using your timer to precisely time this incubation.
- While you are waiting, you can add 1 ml of gelatin to each well of a six-well dish. This should be done in the sterile hood with sterile technique. The gelatin will be removed before you seed the dish with your MES cells but it is important to pre-treat the dish this way. The gelatin must remain in the wells for at least 10 minutes.
- With a 5 ml pipet, add 4 ml of media to the trypsinized MES cells and pipet the liquid up and down (“triturate”) to remove the cells from the plastic and suspend them in the liquid. Remove a small amount of the liquid to an eppendorf tube and take it to the inverted microscopes.
- Fill one chamber of a hemocytometer with 10 ul of the cell suspension. This slide has an etched grid of nine large squares. The square in the center is further etched into 25 squares each with a volume of 0.1 ul and 16 tiny chambers (4x4 pattern). The concentration of cells in a sample can be determined by counting the cells that fall within the 4x4 pattern and then multiplying by 10,000 to determine the number of cells/ml. You should count the cells in the four corner squares of the 25 square grid, then average the numbers to determine the concentration of cells in your suspension.
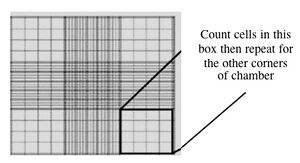
Counting cells using a hemocytometer - You and your partner will seed at different concentrations. Decide if you will try the 1:100 or 1:400 dilution and add the appropriate amount of cell suspension to 10 ml of media in a 15 ml conical tube.
- Remove the gelatin from the six-well dish you have prepared and add 3 ml of your cell dilution to each well. Be sure to label your dish with your name, today’s date, the cell line (called “J1”) and the type of media you have used. Return your cells to the incubator.
- Aspirate any remaining cell suspensions to destroy them and clean up the hood. Dispose any vessels that held cells in the burn box and any sharps in the mayo jars. The next group who uses your hood should find the surfaces wiped down, no equipment left inside, and the sash closed.
- If you are done early, try to calculate the number of cells you'd expect to see next time in each well of your six well dish. Base your calculation on the following three observations:

- only 25% of the cells are able to stick and proliferate (this is called a 25% plating efficiency)
AND
- the doubling time for the cells is 24 hours,
AND
- the cells take 24 hours to recover from trypsin treatment before they begin doubling.
For next week (Due M1D6)
Find the FNW assignment on M1D4.
- Review the tissue culture guidelines in preparation for next time. Next time you set up the culminating experiment of this module!
Reagent list
- Solution I
- 25 mM Tris pH8
- 10 mM EDTA pH8
- 5 mM Glucose
- Solution II
- 1% SDS
- 0.2M NaOH
- Solution III
- 3M KAc, pH 4.8
- several NEB buffers and enzymes available
- see TA notes if interested in media compositions (not needed for methods section)
Navigation Links
Next Day: Mod 1 Day 6: Lipofection Previous Day: Mod 1 Day 4: Ligation & Transformation
