Lidstrom:Enzyme Assay Basics
Back to Protocols
Resources:
- Basics of Enzymatic Assays for HTS. Though written for high-throughput screening, the principles apply to any enzyme assay.
Background
Optimization of Enzyme Assays
Important variables to consider when optimizing a screening assay include: ([1])
- growth conditions
- lysis procedure
- concentrations and volumes of components used in the assay (e.g., enzyme, buffer, substrate)
- the parameter(s) used to identify enzyme function (e.g., kinetics or end-point).
How high should my [enzyme] be?
- The enzyme concentration should usually be no more than 1% of any of small molecules (substrate, cofactors, etc.) in the assay. (0-19-963812-8 pg169)
Assay Validity
From UC Davis ChemWiki An enzyme assay must be considered valid to ensure accurate data and calculations. In order for an enzyme assay to be valid, it must meet multiple requirements:
- Initial rates are being measured.
- V0 must be both reproducible and dependent on [E]t.
- Measuring initial rates demonstrates whether or not the product has reached a substantial concentration.
- Valid assay conditions exist if the product has consumed less than 5% of the substrate.
- pH is constant:
- Enzymes have specific pH ranges of activity due to structural sensitivity to proteins which cause enzymes to be sensitive to pH changes. A prepared buffer solution is used to keep the pH constant so that the pH resist changes.
- Good buffers do not cross membranes; do not aborb light; are chemically stable; and are biochemically inert.
- V0 is proportional to the concentration of the enzyme, [E]t. An assay is only valid when V0 vs [E]t is linear because the enzyme must be the only limiting factor to the substrate concentration. Additionally, when V0 and [E]t are proportional, the presence of effectors are measurable and one can determine if an inhibitor is competitive, noncompetitive, or uncompetitive.
- V0 is corrected for non-enzymatic conversion: V0(corrected) = V0(+enzyme) – V0(-enzyme). A control factor must be measured while conducting enzyme assays in order to ensure accurate calculations. For non-enzyme controls, buffers are used in place of enzymes.
Why rates decline over time
From Enzyme Assays, A Practical Book. By Robert Eisenthal and Michael Danson, 1992. ISBN 0-19-963142-5 or 0-19-963143-3 (paperback). There is also a 2003 edition available @ Amazon & UW libraries.
When you add substrate and enzyme, the rate may be linear for some time then the rate of change may slow. This can be caused by:
- Substrate depletion
- You can test whether this is the case by simply adding more substrate.
- If the initial substrate concentration was much below the Km value, it may be difficult to obtain a prolonged period of linearity unless highly-sensitive assays are used to allow product formation to be detected under conditions where there is a negligible change in substrate concentration.
- Equilibrium
- A reversible reaction may be slowing down because it approaches equilibrium. The rate of backward reaction (P -> S) will increase until the rate of product formation and product consumption are equal.
- You can help this by removing the product as it is formed. A coupled enzyme assay or a reagent that reacts with product may be useful.
- Product inhibition
- Products of enzyme-catalyzed reactions are frequently reversible inhibitors of the reaction. Again, use of a system that removes product is helpful.
- Instability
- Components of the system may break down, including enzymes and reagents.
- How to check:
- Incubate the assay mixture for a series of times under conditions identical to those used in the assay itself but without one of the components (enzyme or substrate(, before starting the reaction by addition of the missing component. If the rates of the reaction are the same whichever component is missing during the pre-incubation period, a loss of linearity due to this cause can probably be excluded.
- It is important to ensure that the conditions of the pre-incubation are identical to the assay itself. For exmaple, many compounds are light sensitive and this can be a particular problem where relatively high intensities of light are used such as is possible in fluorimetry.
- Incubate the assay mixture for a series of times under conditions identical to those used in the assay itself but without one of the components (enzyme or substrate(, before starting the reaction by addition of the missing component. If the rates of the reaction are the same whichever component is missing during the pre-incubation period, a loss of linearity due to this cause can probably be excluded.
- Another problem with optical assays is the use of a narrow slit width resulting in localized destruction of only a small portion of the material in the assay cuvette. For example, the fluorescence of tryptophan solutions may decline with time but removal of the cuvette and shaking it can result in an apparent return to the original level of fluorescence if the photo-destruction is limited to only a small proportion of the total protein.
- It is also possible to for a component in a mixture to appear less' stable under the pre-incubation conditions then when it is in a complete assay mixture. This could result from the substrate being bound by a stabilizing enzyme that protects it from damage from light, etc.
- Time-dependent inhibition
- An enzyme might be less stable when catalyzing the reaction than it is under the pre-incubation conditions described in Section 2.1.4. Such an effect would result in a decline in the rate of the reaction with time, whereas the individual components of the assay mixture might appear quite stable during the pre-incubation experiments.
- To test, you could add more enzyme to the reaction after the reaction ceases. You would expect the rate to be the same as when you started it the first time if the same amount of enzyme is added, unless there has been a significant depletion of substrate(s) or accumulation of inhibitory product(s) during the reaction.
- The enzyme may also generate an inhibitor that binds the enzyme and prevents it from further catalysis.
- You can add more enzyme and see if the rate returns.
- Assay method artefact
- If the specific detection procedure used ceases to respond linearly to increasing product concentrations this can lead to a decline in the measured rate of the reaction with time.
- This can happen with spectrophotometry and fluorometry.
- Coupling enzymes may not respond linearly to product formation over time, for all of the reasons mentioned herein or simply the fact that the coupling system approaching its maximum velocity. Clearly, if such coupled-assay procedures are to be used, it is essential to carry out careful control experiments to prove that the system is capable of providing a true measure of the activity of the enzyme under study at all conditions to be used.
- If the specific detection procedure used ceases to respond linearly to increasing product concentrations this can lead to a decline in the measured rate of the reaction with time.
- Change in assay conditions
- The rate may change when the reaction conditions change.
- Example of such a change: pH changes as rxn progresses. H+ ions are generated or consumed during the course of the reaction and it isn't adequately buffered. You drift from the pH optimum. Check the pH before and after a reaction.
Why & how we measure the initial rate
- Any number of effects can change the velocity of the reaction, so it it best to measure the initial rate, before these factors have their effect.
- If your initial rate is too fast because of substrate depletion or equilibrium, add less enzyme.
- People commonly assume that determining the initial rate when <20% of the substrate is consumed is sufficient to ensure linearity, but this is not garunteed for the reasons in the previous section. Also, this is only valid when the initial substrate concentration is in excess of the Km value.
- You can use polynomial fits to estimate the rate at time = 0.
- Starting an assay by adding one of the components and ensuring adequate mixing can lead to significant uncertainty about the exact time that the reaction was started.
- There may be a burst or lag before the true rate of the reaction is established (section 2.4).
- Note: V is only 91% of Vmax when S is 10 times greater than Km. reference
- Also note, enzymes may also be inhibited at high substrate concentrations.
Bursts and Lags
With assays that show bursts or lag phases, it is important to determine the cause in order to know which phase of the reaction corresponds to the true 'initial rate' of the reaction. The term "initial rate" is normally used to refer to the steady-state rate of the reaction that is established after any pre-steady-state events have occurred.
Potential reasons:
- The temperature may change over time. We usually keep reagents and enzyme preparations on ice, but do the assay at elevated temperatures. Thus there is some change in temperature over time.
- Should we warm up extract/reagent aliquots just before use?
- Particles may settle as the reaction proceeds, leading to erratic rates. This may be interpreted as a burst or lag phase in the reaction.
- To test this: mixing the contents of the assay cuvette after the reaction has become linear should result in a second phase of aberrant behavior.
- Slow detector response
- A lag phase may occur if the initial response of the detection system is slow.
- Slow dissociation of a reversible inhibitor or activator
- Although most reversible enzyme activators and inhibitors will dissociate rapidly from the enzyme when the enzyme-inhibitor mixture is diluted, those that show extremely high affinity for the enzyme may show significant time-dependence in their rates of dissociation from it, and association with it. In such cases dilution of the enzyme-inhibitor mixture into the assay may show a lag as the inhibitor slowly dissociates to its equilibrium value. Conversely, if enzyme is added to a reaction mixture containing inhibitor the rate may slowly decrease until the binding equilibrium has been established.
- Pre-steady-state transients
- It may take time for the concentration of the intermediate enzyme-substrate and enzyme-product complexes to rise to their steady-state levels. Usually this is rapid and takes special equipment to detect.
- Relief of substrate inhibition or activation
- Many enzymes are inhibited by high concentrations of one or more of their substrates. If the initial substrate concentration added to an assay mixture is sufficient to cause some degree of inhibition, the rate of the reaction will tend to increase with time as substrate utilization decreases the inhibition. Alternatively, an initial burst phase n the progress curve can occur if the substrate also behaves as an activator at higher concentrations. Bursts or lags due to such causes should be eliminated by reducing the initial substrate concentration to a level where inhibition, or activation, does not occur. High-substrate inhibition or activation should, of course, be readily detected by their characteristic effects on the dependence of initial velocity on substrate concentration.
- A similar lag phase in the reaction progress curve can occur if the substrate solution is contaminated by a small amount of another substrate for the enzyme which has high affinity for it but which is broken down slowly.
- Activation by product
- A progress curve that curves upward may be observed if one of the products of the reaction is an activator.
- Substrate interconversions
- If a compound exists in more than one form, only one of which is an effective substrate for the enzyme, a slow interconversionb etween these forms can lead to burst or lag phases in the progress curves. Example: sugars in different mutarotated forms.
- Such effects can also give rise to burst phases if a substrate exists in a slow equilibrium between active and inactive forms where an initial rapid phase, corresponding to the utilization of the active form of the substrate, would be followed by a slower phase determined by the rate of isomerization from inactive to active forms.
- Also watch out for substrates that can be in polymeric forms.
- Hysteric effects""
- It is important to eliminate other possible causes of bursts or lags before attributing unusual shaped curves to this. Example: there are two different forms of the enzyme that have different activities.
Blank rates
- It is not uncommon to observe an apparent rate of reaction in the absence of one of the components of the complete assay mixture. You must figure ot the cause in order to make appropriate corrections.
- Try omitting the enzyme and each of the substrates in turn.
Causes:
- Settling of particles
- Especially when lysates have organelles.
- The effect usually slows when particles settle. You can test by re-mixing and see if the rate resumes.
- Precipitation
- Gradual precipitation of material in the assay mixture may lead to similar problems as precipitation.
- You can look for signs of turbidity or visible precipitate formation. You can see if changes in absorbance at wavelengths distant from those where any reaction-dependent changes should occur to monitor changes in turbidity directly.
- Magnesium or calcium ions are added to many assay mixtures because they are essential for the activity of a number of enzymes. However, if such mixtures contain a strong phosphate buffer then precipitation will occur when the solubility product of calcium or magnesium is exceeded.
- A more confusing situation can occur if one of the products of the reaction is not very soluble and precipitates during the later stages of the reaction, giving rise to accelerating progress curves. Although the blank rates arising from precipitation directly affect optical assays, such behavior could also invalidate the results obtained with other assay procedures.
- Gradual precipitation of material in the assay mixture may lead to similar problems as precipitation.
- Contamination of one of the components of the assay mixture
- The presence of one of the substrates in the enzyme solution can give a blank rate with an incomplete reaction mixture. Crude extracts often have some substrate. If the degree of contamination is quite small, the blank rate from this source would be expected to be non-linear and to cease when the endogenous substrate is exhausted.
- If the enzyme is quite stable under the assay conditions it may be possible to wait until the blank rate dies away before starting the assay.
- Alternatively, if the contaminating substrate is a small molecule, it should be possible to remove it by dialysis or gel filtration.
- Problems from this source would be expected to decrease upon purification of the enzyme.
- Some commercially available enzymes contain substrate, which has been added for stability; you can remove it with dialysis or gel filtration.
- It is possible you wouldn't be able to remove contaminants. Examples include gases.
- You may also cause this issue by cross-contamination, such as putting a used pipette in the wrong reagent tube.
- The presence of one of the substrates in the enzyme solution can give a blank rate with an incomplete reaction mixture. Crude extracts often have some substrate. If the degree of contamination is quite small, the blank rate from this source would be expected to be non-linear and to cease when the endogenous substrate is exhausted.
- Adsorption to assay vessels
- Proteins may stick to glass. Enzyme can carry over from one experiment to the next. Distilled water may be insufficient to remove the bound enzyme; an acid wash may be required.
- The use of silicone-treated glass or plastic vessels may minimize this problem but we hav found that not all plastics are inert in this respect. Use disposable cuvettes if suitable for the assay.
- Non-enzymatic reactions
- Solutions of NAD(P)H are unstable at pH values below neutrality, leading to a spontaneous fall in absorbance at 340 nm. Blank rates due to non-enzymatic reactions should be subtracted from the rates given in the presence of enzyme.
- The reaction of exogenous factors can also lead to blank rates. Example: absorption of CO2 in a poorly buffered solution --> lower pH --> blank rate.
- Example: Aldehydes react with NAD+ to give a product that has similar absorbance to NADH. This reaction can cause significant problems in determining the activity of the aldehyde dehydrogenase at alkaline pH values but is not significant at neutral or acid pHs.
- reference = Duncan, R. J. S. and Tipton, K. F. (1971). Eur J. Biochem., 22, 257.
- Contaminating enzymes
- The presence of another enzyme in the preparation which catalyzes an interfering reaction can give rise to a blank rate. If the substrate for the contaminating enzyme is also a contaminant of the preparation it may be possible to remove it by dialysis or gel filtration. However, this is not always possible. For example, the assay of dehydrogenases in crude tissue preparations may be difficult because of the presence of NADH-cytochrome-c reductase. In this case chytochrome-c is not readily removed by dialysis. In such cases it may be necessary to use an inhibitor of the contaminating enzyme, taking care to ensure that it has no effect on the enzyme under study, or to purify the enzyme in order to remove the contaminating material. It may not be sufficient to use an alternative assay procedure if the other enzyme depletes the substrate.
- In some cases, contaminating enzymes may require no substrates other than those used by the enzyme under study.
Correction for blank rates
- It is important to understand the cause of a blank rate before one may make the appropriate corrections for it.
- In many cases it is possible to obtain the true rate of the enzyme-catalyzed reaction by simply subtracting the blank rate given in a suitable incomplete mixture from that obtained with the full assay.
- This approach assumes that:
- the blank rate is an artefact that is unconnected with the activity of the enzyme under study,
- that it continues linearly for the total period of the assay, and that
- it will be unchanged in the full assay.
- In cases where these assumptions are valid, failure to subtract the blank rate will yield apparent anomalies in kinetic behavior.
- If the blank rate occurs in the absence of the enzyme, failure to subtract it will give a plot of initial velocity against enzyme concentration that does not pass through the origin but which shows a finite activity at zero enzyme concentration. Failure to subtract a blank rate that occurs in the absence of one of the substrate can give behavior that does not conform to the Michaelis-Menten equation.
- If an apparent blank rate is due to settling of particles, subtraction of the initial blank rate from the initial rate obtained after starting the reaction may be adequate, but such rates are normally irregular and would be better to await the decline of the blank rate and the stabilization of the assay before measuring the rate. It would be inappropriate to subtract the blank rate if it were due to conamination of the enzyme or the assay mixture contained saturating concentrations of that substrate. In these cases the blank rate is due to the enzyme itself and subtraction would therefore result in an underestimation of the activity.
- A more complicated system where it is inappropriate to subtract an apparent blank rate can occur if an enzyme can catalyze the decomposition of one of its substrates alone but that reaction is supressed by the presence of the second substrate.
- Masking of an assay
- In some cases the activity of a contaminating enzyme may interfere with the assay of an enzyme. The blank rates that can occur in the assay of the enzymes ...
Controls/Blanks
- The basic set of tests you should do:
- + enzymes + substrate
- + enzymes - substrate
- - enzymes + substrate (strain = empty vector control or equivalent)
- - enzymes - substrate (strain = empty vector control or equivalent)
- Mary likes to see a plot like
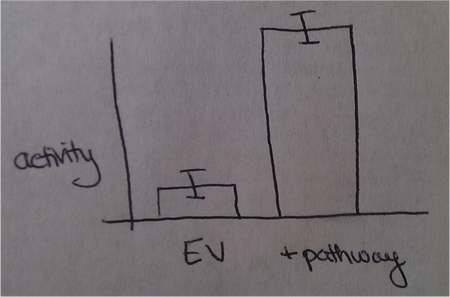
cartoon of a good way to depict enzyme assay data. Each bar is Vmax for reaction with substrate - Vmax for rxn without substrate. - You could also subtract the empty vector height from the control height, but Mary said she prefers to see them separately. JM 10/31/2013

Factors to control for
From Wikipedia 2013/11/1 unless another reference is cited.
- Salt Concentration:
- Most enzymes cannot tolerate extremely high salt concentrations. The ions interfere with the weak ionic bonds of proteins. Typical enzymes are active in salt concentrations of 1-500 mM. As usual there are exceptions such as the halophilic algae and bacteria.
- Effects of Temperature:
- All enzymes work within a range of temperature specific to the organism. Increases in temperature generally lead to increases in reaction rates. There is a limit to the increase because higher temperatures lead to a sharp decrease in reaction rates. This is due to the denaturating (alteration) of protein structure resulting from the breakdown of the weak ionic and hydrogen bonding that stabilize the three dimensional structure of the enzyme active site.[11] The "optimum" temperature for human enzymes is usually between 35 and 40 °C. The average temperature for humans is 37 °C. Human enzymes start to denature quickly at temperatures above 40 °C. Enzymes from thermophilic archaea found in the hot springs are stable up to 100 °C.[12] However, the idea of an "optimum" rate of an enzyme reaction is misleading, as the rate observed at any temperature is the product of two rates, the reaction rate and the denaturation rate. If you were to use an assay measuring activity for one second, it would give high activity at high temperatures, however if you were to use an assay measuring product formation over an hour, it would give you low activity at these temperatures.
- Rates increase by between 4 and 8% per degree C, although at high temperatures denaturation of the enzyme protein decreases product formation. reference
- Effects of pH:
- Most enzymes are sensitive to pH and have specific ranges of activity. All have an optimum pH. The pH can stop enzyme activity by denaturating (altering) the three dimensional shape of the enzyme by breaking ionic, and hydrogen bonds. Most enzymes function between a pH of 6 and 8; however pepsin in the stomach works best at a pH of 2 and trypsin at a pH of 8.
- Substrate Saturation:
- Increasing the substrate concentration increases the rate of reaction (enzyme activity). However, enzyme saturation limits reaction rates. An enzyme is saturated when the active sites of all the molecules are occupied most of the time. At the saturation point, the reaction will not speed up, no matter how much additional substrate is added. The graph of the reaction rate will plateau.
- Level of crowding
- Large amounts of macromolecules in a solution will alter the rates and equilibrium constants of enzyme reactions, through an effect called macromolecular crowding.[13]
- Ionic strength
- This is a complex subject as each enzyme responds in a uniquewayto ionic strength I (salt content).Someenzymes are maximally active at the lowest I, while others require substantial levels of salt for significant activity. Most are inhibited at high (40.5 mol L-1) salt. For most enzymes there is a variation of activity with I, so the value should be fixed and recorded (it is implicit in the total composition of the assay buffer). A natural intracellular ionic strength is typically in the range of 0.15 to 0.2 mol L-1. reference
Assay types
Discontinuous/Stopped assays
To measure the activity of an enzyme one must measure how much product is formed over a given time or, in some cases, how much substrate has been used up, which should be the same thing. Thus ideally a method for measuring either product or substrate in the presence of the other is required. There are many different approaches; this section will deal with what are known as ‘stopped assays’. Stopped assays involve stopping the reaction after a fixed time, then measuring how much product has been formed. Any method is possible, from chemical, enzymatic to bioassay, and generally the simplest is chosen provided it is reliable. In many cases a selective method can distinguish between substrate and product, so that no separation step is required. For example, phosphate release from a phosphate ester can be measured by the standard phosphomolybdate procedure. Otherwise separation of unused substrate from product may be needed. This is essential with radiochemical assays, in which the measurement is of radioactivity, not a specific test for the product itself. Separation methods include chromatographic (thin-layer chromatography, TLC; high-performance liquid chromatography, HPLC), solubility and partition procedures. Methods for stopping the reaction include those which denature the enzyme, such as strong acid, alkali or detergent; heat; or treatments with irreversible inhibitors such as heavy metal ions. In some cases the enzyme can be stopped by addition of a complexing agent such as ethylenediaminetetraacetic acid (EDTA), which removes metal ions essential for activity; even chilling on ice may be sufficient. It is important that stopped assays are checked at least once with varying times of incubation, to ensure that the rate is linear through the period selected for the standard method. reference
- Advantages:
- amenable to higher throughput & autosampling
- Disadvantages:
- You don't get to see the shape of the curve, so impurities and artifacts wont be revealed.
- Timing and volume inaccuracies may be introduced, associated with the termination of the reaction and withdrawing samples at fixed times. (source)
Continuous assays
The alternative to a stopped assay is a continuous one in which the progress of the reaction is followed as it occurs. Continuous assays are much more convenient in that the result is seen immediately, and any deviation the initial rate shows from linearity can be observed. On the other hand not all enzymes have an assay method that can be observed continuously. The simplest continuous assay is one in which the action of the enzyme itself can be followed by changes in absorbance (e.g. NAD(P)H at 340nm with dehydrogenases), fluorescence, viscosity, pH, or one of several other possible physical parameters. In many examples of hydrolase assays, an artificial substrate which releases a coloured or fluorescent product is used. But most enzymes do not produce any change in a readily detectable physical parameter by their activity. This can be overcome using a coupled continuous method. In this process, the product is acted on further (usually by other enzymes that are added to the mixture), until an ultimate product is formed which can be observed physically. A great advantage of coupled assays is that the product is removed, so helping to keep the measured rate constant over a long period by avoiding product inhibition and reversal of reaction. reference
- Advantages:
- You get to see the shape of the curve, so impurities and artifacts are more likely to be revealed.
- Disadvantages:
- Not amenable to high throughput techniques.
Examples

Preparing Samples
Buffer Selection
See Lidstrom:Buffers
Cell Pellet Prep
General guidelines:
- 50 mL of E. coli in LB/TB is usually plenty. (stationary phase)
- If using a methylotroph, you may need more culture volume. Some strains are "sick" and don't grow very turbid. Use:
- 100 mL of at OD 0.6-0.8 ~or~
- 200 mL at OD = 0.4 ~or~
- 300 mL at OD = 0.2
Of course the amount of biomass depends on how well your enzyme is expressed and what its specific activity is.
Lysis
- Use a similar mass of cells for different strains you are breaking.
- Generally you want to keep the samples cold, but also be aware that some proteins denature when they are chilled.
- Make sure you are appropriately buffered. Measure pH before and after lysis.
- Add a reducing compound such as DTT or beta-mercaptoethanol
- Inside cells, the environment is reducing. When you lyse cells, the proteins are exposed to the oxidizing atmosphere and the -SH groups begin to oxidize, causing changes in conformation and activity. By adding reducing agent, you can maintain conditions more similar to the cytosol. beta-mercaptoethanol is cheaper and is usually used ~10-20mM. DTT is more expensive, but is more effective and can be used at ~1mM.
- A little EDTA can increase stability of the proteins by preventing side chains from reacting from heavy metals such as Cu, Pb, Hg, Zn. However, EDTA chealates all metals and may remove metals from the active site of your enzyme!
French Press
- Resuspend in 2 mL of an appropriate lysis buffer
- French press 2-3 times.
- Centrifuge out debris. Perhaps ultracentrifuge.
Sonication
- This has a few disadvantages, and most people only use it when french pressing or other methods are not possible (e.g. Ceci needs to break cells under anaerobic conditions).
- The samples can get quite hot, even when on ice.
- Local high temperatures make free radicals, which can damage proteins. It is vest to keep lysates very concentrated to protect them.
Freeze/Thaw
Enzymatic Lysis
- E.g. Bugbuster
- contains detergents, which may denature proteins
Analyze protein concentration
- 1000 ug/mL is good, says Ceci
Assaying
Concentrations to use
Substrate
- The substrate concentration that is used in enzyme assays is chosen according to parameters such as the Km, the solubility of the substrate, whether high concentrations may inhibit, and the cost of the substrate. reference
Buffer
- See Lidstrom:Buffers
- "An adequate buffer capacity is often only reached at concentrations higher than 25mM. However, hiher buffer concentrations and related ionic strengths can inhibit enzyme activity.. Suitable initial concentrations are therefore between 10 and 25mM. If, after addition of the protein or enzyme, the pH value changes by more than 0.05 units, the concentration of the buffer can first be increased to 50mM. Up to this concentration, no interference was observed with the Good buffers in cell culture experiments." --Applichem booklet
Order of reagents to add
- The most common way it to have a cocktail with all of the components of the reaction except the substrate. You add the key ingredient last. You want to add a large enough volume that the pipetting error is minimized, but a small enough amount to have the temperature consistent. This presumes you have already worked out the correct concentrations of everything for your assay.
- If using a 1mL cuvette, you can:
- add the final reagent as a drop on the side of the cuvette, cover the top with parafilm, and invert a few times to mix.
Volumes to use
- Total volume:
- For the plate reader, Ceci/Amanda always do 200 uL in assays. You can also use less, but then any miniscus effect is relatively more important. The ISBN 0-19-963142-5 says 0.25 mL is best, however you may be limited by expensive substrates/cofactors and having a large reaction volume might represent a trade-off between having number and quality of assays if you are sample limited. Also, the error of the plate reader relative to the signal is maximized at lower ODs. By having a longer path length, your signal to noise ratio increases. ISBN 0-19-963142-5 says that when OD is ~0.2, the error between measurements is 10%.
- Cell extracts:
- Ceci adds 50 uL extract per rxn. She said you don't want to dilute the enzymes more than you need to, as they are most happy when concentrated.
- This may require that you use more substrate. Hopefully your substrate is cheap.
- If you are doing an NADH-linked assay, there is a limit to the amount of NADH you can add, as you will saturate the spectrophotometer.
- Mila uses ~20uL.
- Use little enough that your reaction is slow enough to not miss the fastest part of the reaction. For example, ISBN 0-19-963142-5 says that many scientists experienced with assays often conclude that there is no activity in a sample because they assay with too much extract and the reaction is complete before they start reading the slope.
- Ceci adds 50 uL extract per rxn. She said you don't want to dilute the enzymes more than you need to, as they are most happy when concentrated.
More questions to ask yourself
- Are there other reactions that consume your substrate or intermediates more quickly than the one you are trying to measure?
- If you are testing multiple conditions (such as multiple substrates), you need to consider whether the assay is optimized and validated for each.
Validity Check
- If you reduce the cell extract 2x, do you see Vmax reduce by 2x? (a desired result)
- Note: crowding does change, so your numbers may not be ideal.
- If you double the substrate or cofactor concentration(s), does the rate remain unchanged?
- If the rate changes, it is possible inhibition is occurring. For example, assays that use ATP have ATP degrade to ADP at some rate. If ADP inhibits your assay, then increased in ATP may lead to decreases in Vmax.
Data Processing
Calculating Enzyme Rates
- The slope provided by the plate reader needs to be converted to moles/time/mg protein or similar units.
- Convert absorbance to M
- If using NADH, the extinction coefficient at 340nm is 6220 M-1cm-1
- If there is less volume in the well, the conversion factor is greater because the path length is larger.
- If using NADH, the extinction coefficient at 340nm is 6220 M-1cm-1
- Factor in the path length.
- If using the crystal plate in the plater reader, 200 uL is 0.51 cm. (Elizabeth "Betsy" Skovran figured this out)
- Normalize for protein concentration.
- How to convert units from plate reader to mM/min or uM/min:

Conversion Factors
- (add me!)
Assaying particular compounds
NAD, NADP, NADH, NADPH
- NAD(P)+ absorbs strongly in the ultraviolet due to the adenine base. Peak absorption is at 259 nm, with an extinction coefficient of 16,900 M-1 cm-1 . The reduced forms of NAD(P)H also absorb at a higher wavelength, with a second peak in UV absorption at 339 nm that has an extinction coefficient of 6,220 M-1 cm-1 . This difference in the ultraviolet absorption spectrums between the oxidized and reduced forms of the coenzymes makes it simple to measure the conversion of one to another in enzyme assays by measuring the amount of UV absorption at 339 nm using a spectrophotometer. However, the UV method suffers from poor selectivity, high background and low sensitivity since many biological substances have strong absorption from 250 to 400 nm. It is quite troublesome to use the UV detection method to detect NAD/NADH and NADP/NADPH when they coexist with other biological molecules that have UV absorption from 250 to 400 nm. source

From the 1974 paper: Nicotinamide-Adenine Dinucleotides (NAD, NADP, NADH, NADPH) Spectrophotometric and Fluorimetric Methods. Klingenberg, Martin. Volume 4 (1974), pg2045, retreived from UW library scan 2/2014 JM - NAD and NADP are stable if stored dry and in the dark. Solutions of NAD(P)+ are colorless and stable for about one week at 4 °C with neutral pH, but decompose rapidly in acids. Upon decomposition, they form products that are enzyme inhibitors. (source)

Example of Preparing for an assay that monitors NADH consumption
- First, determine how fast NADH oxidizes in your assay environment. It is pH and buffer dependent. It may not be zero.
- Determine how fast the reaction proceeds in the absence of your enzymes at various substrate concentrations.
- Example: look at Vmax as you increase [formate] for an assay that has formate as a substrate and NADH as the cofactor and substance you are monitoring. You should have an increase in Vmax as you increase [formate] because there are formate dehydrogenases present. As you increase [formate] you will saturate these enzymes (curve 1 in the picture below). You may see that increases in [formate] lead to decreases in Vmax (curve 2 in the cartoon below); this is caused by inhibition. You want to chose a value of formate that is high enough to saturate the background FDHs if you want to observe the rate caused by enzymes you are adding.

Technique Tips
assay tool belt
- test varying [protein]
- add more master mix
- add more of individual components
- heat up sample
logistical tips
- Mark wells you have used to reduce the rate at which you do the wells incorrectly.

Wells that have been used are colored purple or green (on the bottom) to make it clear which column is next. This increases speed and decreases pipetting error frequency. - All plate readers can use 1/2 area plates. This allows you to use half as much sample.
- Bubbles are more of a concern in 1/2 area plates. If you have a tiny bubble in a full-area plate, it doesn't have a huge effect on your measurement. If you have a tiny bubble in a half-area plate, however, the result is almost always noticeable. For this reason, when pipetting liquid into a well of a half-area plate that already contains liquid, don't submerge the pipette tip into the existing liquid. Instead touch it to the side of the plate near the liquid surface. Mixing the plate for 5 seconds will incorporate any liquid that sticks to the side of the well.

avoid bubbles
- Bubbles make the A340 observance of [NADH] look like this:

Misc Tips/Facts
- ATP chealates metal. ISBN 0-19-963142-5 pg 26
- Some enzymes are strongly effected by pH and ionic strength.
- Adding more substrate changes ionic strength, especially if the substrate is charged or multiply charged. ISBN 0-19-963142-5 pg 25-26
- That the optimum pH is reaction direction-specific.
- In more cases than not, the optima in the two directions will be different, especially if there is uptake/ evolution of a proton in the reaction. Some examples of this are with nicotinamide cofactor (NAD(P))-specific dehydrogenases, in which the optimum pH with NAD(P) as oxidizing cofactor is generally in the range 9 to 10, whereas the optimum for reduction of substrate by NAD(P)H is around pH 6 to 7. reference
- Some enzymes lose activity upon dilution
Open Questions
How high can the NADH concentration be before the detector is saturated?
- Will be dependent on the path length.
- Can be tested by making a calibration curve with increasing [NADH]
- NADH is completely saturated at Abs = 3.4, but is most linear at A340 < 1.5. See this page
