Janet B. Matsen:Closed Lab Questions
From OpenWetWare
Jump to navigationJump to search
Back to Janet
I define "closed" as "not bothering me incessantly." One can always learn more but we have to stop at a point otherwise we would never move forward with our research!
If you have any input/corrections/references please drop me a line.
I'm sorry for the low quality pictures. I have a very old phone, and those pics were decently high tech in its day!
Lab Basics:
Why do we disinfect with 70% EtOH? Isn't 100% more potent?
- A: I have read that you don't want to denature the proteins that allow substances like water and ethanol into the cell. If you do, the EtOH can't get into the cytosol. By using 70%, you denature some of the transporter proteins but leave enough to allow more to enter the cell. Also, 70% is cheaper than 100%!
- I still use 100% for everything.
- 70% is still concentrated enough to be very flamable!
How does colony size vary with colony density?
- As the number of colonies per unit area increases, nutrient diffusion becomes limiting. New molecular biologists are sometimes worried that variation in colony sizes on a plate indicates contamination, but it can be caused by tight packing.
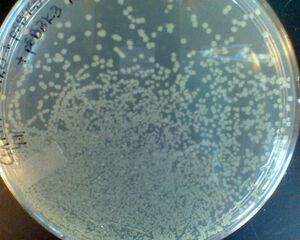
colony size varies with colony density

Culture Media
Wow.. these 2 batches of TB media are very different in color... did I mess up?
- A:Maybe, but not necessarily. Different batches of yeast and tryptone can have very different compositions. This is one of the purposes of defined media.

these two TB batches were made with the same recipe but different bottles of one of the yeast-derived reagents. I forget whether it was yeast extract or tryptone.

What is the difference between agar and agarose?
- "Agar is a heterogeneous mixture of two classes of polysaccharide: agaropectin and agarose. Although both polysaccharide classes share the same galactose-based backbone, agaropectin is heavily modified with acidic side-groups, such as sulfate and pyruvate. The neutral charge and lower degree of chemical complexity of agarose make it less likely to interact with biomolecules, and, therefore, agarose has become the preferred matrix for work with proteins and nucleic acids." (source)
- Note: some bacteria can grow by eating agarose!
Why do we grow E. coli on glycerol, not glucose?
- Amanda Smith found:
- "Glycerol play the part of a cheap carbon-energy source for E.coli.
- Ecoli grown on glycerol excretes less acetate compared to e coli grown on glucose. (due to changes in the biochemical pathways and slower absorption rate of glycerol). Acetate is bad because it limits the growth of e coli. This is due primarily by methionine synthesis inhibition, which in turn disrupts protein synthesis and alters protein expression (A horror story if you are expressing proteins). Acetate also acidifies the media which also cause other changes in E coli physiology (such as reduced growth rate, altered K/Na ion and amino acid anions concentration. Which also has effects on protein synthesis)"
- "Glycerol play the part of a cheap carbon-energy source for E.coli.
Colony PCR
The gel is dark where I loaded it -- what does this mean?
- A: It probably means you overloaded the PCR with too many cells.
What causes an overloaded colony PCR reaction to fail?
- A: Thermostable polymerases require magnesium (link). It has been suggested that because DNA chealates magnesium, overloading a colony PCR may inhibit the polymerase. (11/2012)
- I'd love to test that by titrating in extra Mg in a series of overloaded colony PCRs. It is also possible there is a crowding effect or unfavorable interactions with the cell lysate and the polymerase.
Primers/Oligos
Should I pay extra to further purify large bp oligos?
- Usually no. The sequencing companies just want your money. Do sequence your creations to confirm that you made what you expected and remember you can always screen a new colony if the first one looks bad.
My primers are yellow!

- Don't worry -- this is normal! Moreover, it is extra visible when you order a lot of a primer (large synthesis scale) because there is more of it to see.
Cloning
How do I decide whether to gel purify after digestions (before ligations)
- The BioBrick/Ginkgo manual doesn't instruct you to. Besty always does.
- Um... I don't do much restriction enzyme cloning at this point... Gibson rules! 7/2012
How does oxygen limitation cause lower plasmid copy number? What about the growth medium?
Healthy/happy cells --> more protein or DNA. It's just a fact.
- TB gives way higher yield than LB
- Growing cultures with liquid:air volumes higher than 1:5 is very bad for plasmid yield.
Transformations
Is it ok to re-use electroporation cuvettes after they arc and turn black?

- Electroporation cuvettes are aluminum. The "black stuff" that appears after arcing is aluminum oxide. Though this usually appears low in the cuvette away from the pocket that holds the cells, it correlates to oxidation & non-uniformity in the metal that does contact the cells. This non-uniformity makes arcing more likely when you electroporate a future sample. You will notice that re-used cuvettes are more likely to arc, and that ones with visible oxidation are even more likely to arc.
- To reduce arcing, reduce the resistance (ohms) setting. Though you do need some current to flow through the cells for electroporation, reduced current leads to less arcing. I set my minimum to 200 ohms & maximum to 500 ohms 2012/07/31.
- To avoid arcing completely, use chemically competent cells instead. Though it is more work to prepare the special growth media, you will save time/money/DNA reletive to electroporation.
- added 7/31/2012
How does the density of electrocompetent cells affect transformation efficiency?

- It matters a LOT! The undiluted samples (dilution factor = 0) were harvested ad mid-exponential phase and resuspended in 1/500ths of the culture volume. Details of my experiment are here.
What is the ballpark efficiency of electroporation?
- In the experiment described just above, I calculated that for 0.1 ng of plasmid, the maximum efficiency was 5*10-6, defined as the fraction of plasmids that led to colonies (not CFU/ug DNA). I haven't looked up literature values to see how this compares to published statistics. This statistic makes Gibson cloning seem even more amazing. Even if you get only a few colonies, there were likely many orders of magnitudes more viable plasmids in the transformation product than that!
Is it important to let chemically competent cells sit on ice 20-30 min after adding DNA? What does this do? (7/13/2012)
- Example protocol showing ice incubation
- Mila weighing in: "This waiting period is part of the protocol. Not sure anyone knows what exactly happens but likely DNA just needs this time to land onto a cell. Longer time does not do much for the efficiency of the transformation. However the efficiency of the cells is determined by something upstream, by how the cells were grown and chemically treated. This is the most important part of the transformation process.."
- online: ""After making the cells leaky, the DNA is added to the cells and allowed to sit on ice for 10-20 minutes. This allows the DNA to get past the cell membrane, and gives enough time for lots of cells to recieve the DNA and to make certain all of the DNA gets in the cells. After this, in order to make the cells keep the DNA, and to make certain they survive, (Being leaky is not a good thing) the cells are heat shocked for several seconds to 'turn on' (induce) heat shock genes which aid in survival and recovery. After that, the cells are incubated to start growing and plated on selective media to recover those cells that actually recieved the DNA.
- It is simple to do an experiment where you transform some without waiting and some after waiting, of course.
Gene Expression & Plasmids
Is a two plasmid system where the two plasmids have different antibiotic resistance genes but the same origin of replication stable?
- A collaborator implied use of a different antibiotic is sufficient. However, I suppose this could lead to unpredictability in the number of each plasmid. For example, suppose the copy origin they share yields 100 plasmid copies. It could be that 10 copies of the antibiotic A & 90 copies of the antibiotic B plasmids is equally favorable as 90 copies of the antibiotic A plasmid & 10 of the antibiotic B plasmid. Presuming the two plasmids encode different proteins, the relative ratios of the expression levels would be drastically important.
- If allowed to compete in a flask, the burden of each plasmid will affect the distribution of the plasmids.
- added 7/16/2012 after talking to BioRad customer service
BioBricks
The parts in the registry have sequence that don't include the prefix & suffix... do they come with it?
- Yes, they come with a prefix/suffix. You can find which BioBrick method a part is compatable with under the part's "Assembly Compatibility" listing. Most are RFC 10 (http://partsregistry.org/partsdb/scars.cgi), the standard format.
Buffers
How do method developer chose between sodium and potassium phospahte buffers in an assay? 5/2013
- Facts:
- The pKas for sodium and potassium phosphate salts are the same.
- The buffering capacities are the same.
- Potential issues:
- Na or K interferance with an enzyme
- It is possible one will interfere more than the other.
- Solubility
- Sometimes Na precipitates when K wouldn't & vice versa
- Na or K interferance with an enzyme
- Michael Konopka's words: "What assays are you trying to measure? As for the buffering capacity, that really shouldn't matter since the pertinent components for the buffer (mono- and di-basic phosphate) are the same. The issue is if the potassium or sodium ion will form a precipitate which you don't want around if mixing with other solutions (or do want). A classic example is potassium will precipitate SDS while sodium is soluble. That's why in minipreps one adds potassium acetate/acetic acid after using SDS/NaOH to lyse the cells/dissolve lipids & proteins. The proteins, lipids, and chromosomal DNA is then trapped in the precipitate (plasmid DNA still in solution)."
What is the different between phosphate buffer & PBS (phosphate buffered sailine)?
- Phosphate buffered saline has MUCH lower concentration of phospate buffer. It is mostly salt.
How stable is HEPES?
- Not stable. Make it the day you use it.
How do I decide the spacing between a promoter and ribosome binding site? A ribosome binding site & the start codon?
- Recall that a promoter starts making a transcript at the +1 site, no matter the sequence, and that the promoter is only for transcription and the RBS is only for translation. The -10 and -35 locations in the promoter that are significant for binding are numbered relative to this +1 site. You can have any arbitrary sequence before the RBS, and people often include operators for genetic regulation. Or, you can put the ribosome binding site next. This is more standard. The ribosome binding site needs to be correctly spaced from the start codon, and you have to look at the individual RBS to determine it. For some, a BioBrick scar is the perfect distance between the end of the RBS and the start codon; this is useful for making an expression vector like UW's 2010 iGEM vectors.
Miscellaneous
Is it "media" or "medium"?
I have recently switched to using "medium" when describing a single type of solid or liquid growth substance.
