Dahlquist:Microarray Data Analysis Workflow

This is the most current version of the data analysis protocol for the Dahlquist Lab microarray data.
- To access the version that was used during Week 1 of SURP 2015, follow this link.
- During SURP 2019, we are validating that each section of these instructions is up-to-date. A comment will be made to the code of each section as it is validated.
- This workflow is used with a DNA microarray dataset submitted to the NCBI GEO repository as series GSE83656.
Summary of steps for microarray data analysis
- Quantitate the fluorescence signal in each spot (GenePix Pro)
- Calculate the ratio of red/green fluorescence (GenePix Pro)
- Log transform the ratios (GenePix Pro)
- Normalize the ratios on each microarray slide (within-chip normalization)
- Normalize the ratios for a set of slides in an experiment (between-chip normalization)
- Perform statistical analysis on the ratios
- Within-strain ANOVA
- Modified t test for each timepoint
- Between-strain ANOVA
- Benjamini & Hochberg and Bonferroni p value corrections for the above three tests
- "Sanity Check" on above three tests
- Pattern finding algorithms (clustering with stem)
- Gene Ontology term enrichment analysis (on clusters with stem or on gene sets with MAPPFinder)
- Pathway analysis (GenMAPP)
- Determining candidate transcription factors and gene regulatory network (YEASTRACT)
- Dynamical modeling with GRNmap; visualization with GRNsight
Before you begin...
You will record all of the manipulations of the data in an electronic lab notebook stored on this wiki. Please see the Wiki Checklist page for more details on how to do this.
Viewing File Extensions
- The Windows 7 and Windows 10 operating systems defaults to hiding file extensions. To turn them back on, do the following:
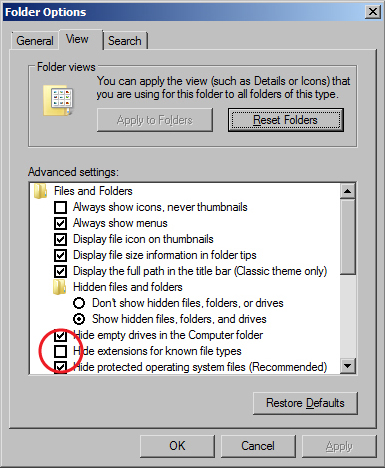
Folder Options window - In Windows 7, do the following:
- Go to the Start menu and select "Control Panel".
- In the window that appears, search for "Folder Options" in the search field in the upper right hand corner.
- Click on "Folder Options" in the main window.
- When the Folder Options window appears, click on the View tab.
- Uncheck the box for "Hide extensions for known file types".
- Click the OK button.
- In Windows 10, do the following:
- Go to Cortana (the search function), a circle icon to the right of the start menu and search for "File Explorer Options".
- Open File Explorer Options in the search results.
- When the File Explorer Options window appears, click on the View tab.
- Uncheck the box for "Hide extensions for known file types".
- Click the OK button.
- Note that the computers in the Seaver 120 computer lab and other labs on campus are are set to erase all custom user settings and restore the defaults once they have been restarted, so you will probably have to turn on file extensions multiple times when using the lab computers. The Dahlquist Lab computers do not erase the settings, so this step only needs to be performed once.

Set Your Browser to Prompt You for the Location to Save your Downloaded Files
- In Mozilla Firefox, open the Options window.
- Select the radio button that says "Always ask me where to save files".
- You could also change the default "Save files to" location to your Desktop, so that will be the first choice when it prompts you where to save the file. (You will have to temporarily deselect the radio button to do this and then reselect it when you are done.
- Click OK to save your changes.
- In Google Chrome, open the Settings window.
- Click on the link at the bottom of the page that says "Advanced Settings".
- Check the box that says "Ask where to save each file before downloading".
- You could also change the default Download location to your Desktop, so that will be the first choice when it prompts you where to save the file.
- Your settings are automatically saved.
Steps 1-3: Generating Log2 Ratios with GenePix Pro
- The protocol for gridding and generating the intensity (log2 ratio) data with GenePix Pro 6.1 is found on this page.
- This protocol will generate a
*.gprfile for each chip which is then fed into the normalization protocol below. - To bypass this step, you can download the
*.gprfiles from the NCBI GEO repository series GSE83656. Click this link to begin the download of a 154.9 Mb compressed file called "GSE83656_RAW.tar".
Steps 4-5: Within- and Between-chip Normalization
- The protocol supplants the one found on this page.
- The scripts and accessory files required to run the normalization are archived on the Dahlquist Lab GitHub Repository.
Installing R x64 v3.1.0 and the limma v3.20.1 package
The following protocol was developed to normalize GCAT and Ontario DNA microarray chip data from the Dahlquist Lab using the R Statistical Software and the limma package (part of the Bioconductor Project).
Note that if R x64 v3.1.0 and the limma v3.20.1 package are already installed on your computer, you can proceed to the next section.
- The normalization procedure has been verified to work with the 64-bit (x64) version 3.1.0 of R released in April 2014 (link to download site) and and version 3.20.1 of the limma package (direct link to download zipped file]) on the Windows 7 platform.
- Note that using other versions of R or the limma package might give different results.
- Note also that using the 32-bit versus the 64-bit versions of R 3.1.0 will give different results for the normalization out in the 10-13 or 10-14 decimal place. The Dahlquist Lab has standardized on using the 64-bit version of R.
- To install R for the first time, download and run the installer from the link above, accepting the default installation.
- To use the limma package, unzip the file and place the contents into a folder called "limma" in the library directory of the R program. If you accept the default location, that will be C:\Program Files\R\R-3.1.0\library (this will be different on the computers in S120 since you do not have administrator rights).
Running the Normalization Script
This protocol describes the procedure for normalizing the microarray data for the wild type and transcription factor deletion strains Δcin5, Δgln3, Δhap4, Δhmo1, Δswi4, and Δzap1 which were hybridized on the GCAT and Ontario chip types. It can also be used to normalize the Saccharomyces paradoxus data, which was hybridized to the Ontario chips.
- Create a folder on your Desktop to store your files for the microarray analysis procedure.
- Download the zipped file that contains the
.gprfiles and save it to this folder (or move it if it saved in a different folder).- Unzip this file using 7-zip. Right-click on the file and select the menu item, "7-zip > Extract Here".
- Download the following files by right-clicking on the link and choosing the menu item "Save Link As..."
- Download the GCAT_Targets_20160616.csv file and Ontario_Targets_wt-dCIN5-dGLN3-dHAP4-dHMO1-dSWI4-dZAP1_20160616.csv file and save them to this folder (or move them if they saved to a different folder).
- Download the GCAT-and-Ontario_normalization_script.R script and save (or move) it to this folder.
- Download the generate_MA_and_box_plots.R script and save (or move) it to this folder.
- Note that the filenames for the GCAT and Ontario Targets files and the strains are hardcoded into the normalization script. They can be modified for use with other data by modifying the appropriate lines in the script.
- Launch R x64 3.1.0 (make sure you are using the 64-bit version).
- Change the directory to the folder containing the targets file and the
*.gprfiles for the chips by selecting the menu item File > Change dir... and clicking on the appropriate directory. You will need to click on the + sign to drill down to the right directory. Once you have selected it, click OK. - In R, select the menu item File > Source R code..., and select the GCAT-and-Ontario_normalization_script.R script.
- Wait while R processes your files.
- When the processing has finished, you will find three files called GCAT_and_Ontario_Unnormalized.csv, GCAT_and_Ontario_Within_Array_Normalization.csv, GCAT_and_Ontario_Between_Array_Normalization.csv. The latter file is the one that you will need going forward.
- Save back-ups of these files to to a flash drive.
- Note that the GCAT_and_Ontario_Within_Array_Normalization.csv file is the source of, and should be identical to the processed data submitted to NCBI GEO as series GSE83656.
Visualizing the Normalized Data
- Immediately after running the normalization script, select the menu item File > Source R code..., and select the generate_MA_and_box_plots.R script.
- Wait while R processes your files. You will see the individual plots being created in a new window. R will save them automatically to the same folder that contains the data and scripts.
- The box plots for each strain (comparison of the before, after within- and after between-chip normalization in the same file) are saved under the name of the strain, e.g., dCIN5.jpg.
- The MA plots are saved under a name for the individual chip, e.g., dCIN5_LogFC_t15-1.jpg, and show the plots both before and after normalization.
- Wait while R processes your files. You will see the individual plots being created in a new window. R will save them automatically to the same folder that contains the data and scripts.
- Zip the files of the plots together and save back-up copies of them to a flash drive.
Step 6: Statistical Analysis
Preparing the Excel Workbook
- For the statistical analysis, we will begin with the file "GCAT_and_Ontario_Between_Array_Normalization.csv" that you generated in the previous step.
- Open this file in Excel and Save As an Excel Workbook
*.xlsx. It is a good idea to add your initials and the date (yyyymmdd) to the filename as well. - Rename the worksheet with the data "Master_Sheet".
- Type the header "ID" in cell A1.
- Insert a new column after column A and name it "Standard Name". Column B will contain the common names for the genes on the microarray.
- Copy the entire column of IDs from Column A.
- Paste the names into the "Value" field of the ORF List <-> Gene List tool in YEASTRACT. Then, click on the "Transform" button.
- Select all of the names in the "Gene Name" column of the resulting table.
- Copy and paste these names into column B of the
*.xlsxfile. Save your work.
- Insert a new column on the very left and name it "MasterIndex". We will create a numerical index of genes so that we can always sort them back into the same order.
- Type a "1" in cell A2 and a "2" in cell A3.
- Select both cells. Hover your mouse over the bottom-right corner of the selection until it makes a thin black + sign. Double-click on the + sign to fill the entire column with a series of numbers from 1 to 6189 (the number of genes on the microarray).
- Some cells in the worksheet have "NA" either because there was missing data for genes that existed on the Ontario chips, but not the GCAT chips or because that spot was flagged as "Not Found" by the GenePix Pro software.
- Select the menu item Find/Replace and Find all cells with "NA" and replace them with a single space character. Record how many replacements were made to your electronic lab notebook. Save your work.
- This will be the starting point for our statistical analysis below.
Within-strain ANOVA
The purpose of the witin-stain ANOVA test is to determine if any genes had a gene expression change that was significantly different than zero at any timepoint.
- Create a new worksheet, naming it either "(STRAIN)_ANOVA" as appropriate. For example, you might call yours "wt_ANOVA" or "dHAP4_ANOVA"
- Copy the first three columns containing the "MasterIndex", "ID", and "Standard Name" from the "Master_Sheet" worksheet for your strain and paste it into your new worksheet. Copy the columns containing the data for your strain and paste it into your new worksheet.
- At the top of the first column to the right of your data, create five column headers of the form (STRAIN)_AvgLogFC_(TIME) where (STRAIN) is your strain designation and (TIME) is 15, 30, etc.
- In the cell below the (STRAIN)_AvgLogFC_t15 header, type
=AVERAGE( - Then highlight all the data in row 2 associated with (STRAIN) and t15, press the closing paren key (shift 0),and press the "enter" key.
- This cell now contains the average of the log fold change data from the first gene at t=15 minutes.
- Click on this cell and position your cursor at the bottom right corner. You should see your cursor change to a thin black plus sign (not a chubby white one). When it does, double click, and the formula will magically be copied to the entire column of 6188 other genes.
- Repeat steps (4) through (8) with the t30, t60, t90, and the t120 data.
- Now in the first empty column to the right of the (STRAIN)_AvgLogFC_t120 calculation, create the column header (STRAIN)_ss_HO.
- In the first cell below this header, type
=SUMSQ( - Highlight all the LogFC data in row 2 for your (STRAIN) (but not the AvgLogFC), press the closing paren key (shift 0),and press the "enter" key.
- In the next empty column to the right of (STRAIN)_ss_HO, create the column headers (STRAIN)_ss_(TIME) as in (3).
- Make a note of how many data points you have at each time point for your strain. For most of the strains, it will be 4, but for dHAP4 t90 or t120, it will be "3", and for the wild type it will be "4" or "5". Count carefully. Also, make a note of the total number of data points. Again, for most strains, this will be 20, but for example, dHAP4, this number will be 18, and for wt it should be 23 (double-check).
- In the first cell below the header (STRAIN)_ss_t15, type
=SUMSQ(<range of cells for logFC_t15>)-COUNTA(<range of cells for logFC_t15>)*<AvgLogFC_t15>^2and hit enter.- The phrase <range of cells for logFC_t15> should be replaced by the data range associated with t15.
- The
COUNTAfunction counts the number of cells with data points in the range specified (i.e., it doesn't count cells with missing values). - The phrase <AvgLogFC_t15> should be replaced by the cell number in which you computed the AvgLogFC for t15, and the "^2" squares that value.
- Upon completion of this single computation, use the Step (7) trick to copy the formula throughout the column.
- Repeat this computation for the t30 through t120 data points. Again, be sure to get the data for each time point, type the right number of data points, and get the average from the appropriate cell for each time point, and copy the formula to the whole column for each computation.
- In the first column to the right of (STRAIN)_ss_t120, create the column header (STRAIN)_SS_full.
- In the first row below this header, type
=sum(<range of cells containing "ss" for each timepoint>)and hit enter. - In the next two columns to the right, create the headers (STRAIN)_Fstat and (STRAIN)_p-value.
- Recall the number of data points from (13): call that total n.
- In the first cell of the (STRAIN)_Fstat column, type
=((n-5)/5)*(<(STRAIN)_ss_HO>-<(STRAIN)_SS_full>)/<(STRAIN)_SS_full>and hit enter.- Don't actually type the n but instead use the number from (13). Also note that "5" is the number of timepoints and the dSWI4 strain has 4 timepoints (it is missing t15).
- Replace the phrase (STRAIN)_ss_HO with the cell designation.
- Replace the phrase <(STRAIN)_SS_full> with the cell designation.
- Copy to the whole column.
- In the first cell below the (STRAIN)_p-value header, type
=FDIST(<(STRAIN)_Fstat>,5,n-5)replacing the phrase <(STRAIN)_Fstat> with the cell designation and the "n" as in (13) with the number of data points total. (Again, note that the number of timepoints is actually "4" for the dSWI4 strain). Copy to the whole column. - Before we move on to the next step, we will perform a quick sanity check to see if we did all of these computations correctly.
- Click on cell A1 and click on the Data tab. Select the Filter icon (looks like a funnel). Little drop-down arrows should appear at the top of each column. This will enable us to filter the data according to criteria we set.
- Click on the drop-down arrow on your (STRAIN)_p-value column. Select "Number Filters". In the window that appears, set a criterion that will filter your data so that the p value has to be less than 0.05.
- Excel will now only display the rows that correspond to data meeting that filtering criterion. A number will appear in the lower left hand corner of the window giving you the number of rows that meet that criterion. We will check our results with each other to make sure that the computations were performed correctly.
Calculate the Bonferroni and p value Correction
- Now we will perform adjustments to the p value to correct for the multiple testing problem. Label the next two columns to the right with the same label, (STRAIN)_Bonferroni_p-value.
- Type the equation
=<(STRAIN)_p-value>*6189, Upon completion of this single computation, use the Step (10) trick to copy the formula throughout the column. - Replace any corrected p value that is greater than 1 by the number 1 by typing the following formula into the first cell below the second (STRAIN)_Bonferroni_p-value header:
=IF(<STRAIN_Bonferroni_p-value>1,1,<STRAIN_Bonferroni_p-value>). Replace <STRAIN_Bonferroni_p-value> with the cell designation where that value was computed. Use the Step (10) trick to copy the formula throughout the column.
Calculate the Benjamini & Hochberg p value Correction
- Insert a new worksheet named "(STRAIN)_ANOVA_B-H".
- Copy and paste the "MasterIndex", "ID", and "Standard Name" columns from your previous worksheet into the first two columns of the new worksheet.
- For the following, use Paste special > Paste values. Copy your unadjusted p values from your ANOVA worksheet and paste it into Column D.
- Select all of columns A, B, C, and D. Sort by ascending values on Column D. Click the sort button from A to Z on the toolbar, in the window that appears, sort by column D, smallest to largest.
- Type the header "Rank" in cell E1. We will create a series of numbers in ascending order from 1 to 6189 in this column. This is the p value rank, smallest to largest. Type "1" into cell E2 and "2" into cell E3. Select both cells E2 and E3. Double-click on the plus sign on the lower right-hand corner of your selection to fill the column with a series of numbers from 1 to 6189.
- Now you can calculate the Benjamini and Hochberg p value correction. Type (STRAIN)_B-H_p-value in cell F1. Type the following formula in cell F2:
=(D2*6189)/E2and press enter. Copy that equation to the entire column. - Type "STRAIN_B-H_p-value" into cell G1.
- Type the following formula into cell G2:
=IF(F2>1,1,F2)and press enter. Copy that equation to the entire column. - Select columns A through G. Now sort them by your MasterIndex in Column A in ascending order.
- Copy column G and use Paste special > Paste values to paste it into the next column on the right of your ANOVA sheet.
- Upload the .xlsx file that you have just created to LionShare. Send Dr. Dahlquist an e-mail with the link to the file (e-mail kdahlquist at lmu dot edu).
Sanity Check: Number of genes significantly changed
Before we move on to further analysis of the data, we want to perform a more extensive sanity check to make sure that we performed our data analysis correctly. We are going to find out the number of genes that are significantly changed at various p value cut-offs.
- Go to your (STRAIN)_ANOVA worksheet.
- Select row 1 (the row with your column headers) and select the menu item Data > Filter > Autofilter (The funnel icon on the Data tab). Little drop-down arrows should appear at the top of each column. This will enable us to filter the data according to criteria we set.
- Click on the drop-down arrow for the unadjusted p value. Set a criterion that will filter your data so that the p value has to be less than 0.05.
- How many genes have p < 0.05? and what is the percentage (out of 6189)?
- How many genes have p < 0.01? and what is the percentage (out of 6189)?
- How many genes have p < 0.001? and what is the percentage (out of 6189)?
- How many genes have p < 0.0001? and what is the percentage (out of 6189)?
- When we use a p value cut-off of p < 0.05, what we are saying is that you would have seen a gene expression change that deviates this far from zero by chance less than 5% of the time.
- We have just performed 6189 hypothesis tests. Another way to state what we are seeing with p < 0.05 is that we would expect to see this a gene expression change for at least one of the timepoints by chance in about 5% of our tests, or 309 times. Since we have more than 309 genes that pass this cut off, we know that some genes are significantly changed. However, we don't know which ones. To apply a more stringent criterion to our p values, we performed the Bonferroni and Benjamini and Hochberg corrections to these unadjusted p values. The Bonferroni correction is very stringent. The Benjamini-Hochberg correction is less stringent. To see this relationship, filter your data to determine the following:
- How many genes are p < 0.05 for the Bonferroni-corrected p value? and what is the percentage (out of 6189)?
- How many genes are p < 0.05 for the Benjamini and Hochberg-corrected p value? and what is the percentage (out of 6189)?
- In summary, the p value cut-off should not be thought of as some magical number at which data becomes "significant". Instead, it is a moveable confidence level. If we want to be very confident of our data, use a small p value cut-off. If we are OK with being less confident about a gene expression change and want to include more genes in our analysis, we can use a larger p value cut-off.
- Comparing results with known data: the expression of the gene NSR1 (ID: YGR159C)is known to be induced by cold shock. Find NSR1 in your dataset. What is its unadjusted, Bonferroni-corrected, and B-H-corrected p values? What is its average Log fold change at each of the timepoints in the experiment? Note that the average Log fold change is what we called "STRAIN)_AvgLogFC_(TIME)" in step 3 of the ANOVA analysis.
- We will compare the numbers we get between the wild type strain and the other strains studied, organized as a table. Use this sample PowerPoint slide to see how your table should be formatted.
Modified t test for each timepoint
In the analysis above we performed an ANOVA to determine if any genes had a gene expression change that was significantly different than zero at any timepoint. Now we will perform a modified t test to determine if any genes had a gene expression change that was significantly different than zero at each timepoint. You will perform your analysis on the same strain that you did above, adding these calculations to the same Excel workbook.
- Insert a new worksheet into your Excel workbook and name it "(STRAIN)_ttest", e.g., "wt_ttest" or "dHAP4_ttest".
- Go back to the "Master_Sheet" worksheet for your strain. Copy the first three columns containing the "MasterIndex", "ID", and "Standard Name" from the "Master_Sheet" worksheet for your strain and paste it into your new worksheet. Copy the columns containing the data for your strain and paste it into your new worksheet.
- Go to the empty columns to the right on your worksheet. Create new column headings in the top cells to label the average log fold changes that you will compute. Name them with the pattern <dHAP4>_<AvgLogFC>_<tx> where you use the appropriate text within the <> and where x is the time. For example, "dHAP4_AvgLogFC_t15".
- Compute the average log fold change for the replicates for each timepoint by typing the equation:
=AVERAGE(range of cells in the row for that timepoint)
into the second cell below the column heading. For example, your equation might read
=AVERAGE(C2:F2)
Copy this equation and paste it into the rest of the column.
- Create the equation for the rest of the timepoints and paste it into their respective columns. Note that you can save yourself some time by completing the first equation for all of the averages and then copy and paste all the columns at once.
- Go to the empty columns to the right on your worksheet. Create new column headings in the top cells to label the T statistic that you will compute. Name them with the pattern <dHAP4>_<Tstat>_<tx> where you use the appropriate text within the <> and where x is the time. For example, "dHAP4_Tstat_t15". You will now compute a T statistic that tells you whether the normalized average log fold change is significantly different than 0 (no change in expression). Enter the equation into the second cell below the column heading:
=AVERAGE(range of cells)/(STDEV(range of cells)/SQRT(number of replicates))
For example, your equation might read:
=AVERAGE(C2:F2)/(STDEV(C2:F2)/SQRT(4))
(NOTE: in this case the number of replicates is 4. Be careful that you are using the correct number of parentheses.) Copy the equation and paste it into all rows in that column. Create the equation for the rest of the timepoints and paste it into their respective columns. Note that you can save yourself some time by completing the first equation for all of the T statistics and then copy and paste all the columns at once.
- Go to the empty columns to the right on your worksheet. Create new column headings in the top cells to label the P value that you will compute. Name them with the pattern <dHAP4>_<Pval>_<tx> where you use the appropriate text within the <> and where x is the time. For example, "dHAP4_Pval_t15". In the cell below the label, enter the equation:
=TDIST(ABS(cell containing T statistic),degrees of freedom,2)
For example, your equation might read:
=TDIST(ABS(AE2),3,2)
The number of degrees of freedom is the number of replicates minus one. Copy the equation and paste it into all rows in that column.
- As with the ANOVA, we encounter the multiple testing problem here as well.
Bonferroni Correction
- Now we will perform adjustments to the p value to correct for the multiple testing problem. Label the columns to the right with the label, (STRAIN)_Bonferroni-Pval_tx (do this twice in a row).
- Type the equation
=<(STRAIN)_Pval_tx>*6189, Upon completion of this single computation, use the trick to copy the formula throughout the column. - Replace any corrected p value that is greater than 1 by the number 1 by typing the following formula into the first cell below the second (STRAIN)_Bonferroni-Pval_tx header:
=IF(r2>1,1,r2). Use the trick to copy the formula throughout the column.
Benjamini & Hochberg Correction
- Insert a new worksheet named "(STRAIN)_ttest_B-H". You will need to perform the procedure below for the p values for each timepoint. Do them individually one at a time to avoid confusion.
- Copy and paste the "MasterIndex", "ID", and "Standard Name" columns from your previous worksheet into the first two columns of the new worksheet.
- For the following, use Paste special > Paste values. Copy your unadjusted p values from the first timepoint from your ttest worksheet and paste it into Column D.
- Select all of columns A, B, C, and D. Sort by ascending values on Column D. Click the sort button from A to Z on the toolbar, in the window that appears, sort by column D, smallest to largest.
- Type the header "Rank" in cell E1. We will create a series of numbers in ascending order from 1 to 6189 in this column. This is the p value rank, smallest to largest. Type "1" into cell E2 and "2" into cell E3. Select both cells E2 and E3. Double-click on the plus sign on the lower right-hand corner of your selection to fill the column with a series of numbers from 1 to 6189.
- Now you can calculate the Benjamini and Hochberg p value correction. Type (STRAIN)_B-H_Pval_tx in cell F1. Type the following formula in cell F2:
=(D2*6189)/E2and press enter. Copy that equation to the entire column. - Type "STRAIN_B-H_Pval_tx" into cell G1.
- Type the following formula into cell G2:
=IF(F2>1,1,F2)and press enter. Copy that equation to the entire column. - Select columns A through G. Now sort them by your MasterIndex in Column A in ascending order.
- Copy column G and use Paste special > Paste values to paste it into the next column on the right of your ttest sheet.
- Upload the .xlsx file that you have just created to LionShare. Note that when you upload your file, you should check the box to "Overwrite file if it already exists." This will then replace your previous version of your file with the updated one containing today's calculations. Send Dr. Dahlquist an e-mail with the link to the file (e-mail kdahlquist at lmu dot edu).
Sanity Check
- We will also perform the "sanity check" as follows:
- Determine how many genes have a p value < 0.05 at each timepoint.
- Keeping the "Pval" filter at p < 0.05, How many have an average log fold change of > 0.25 and p < 0.05 at each timepoint? How many have an average log fold change of < -0.25 and p < 0.05 at each timepoint? (These log fold change cut-offs represent about a 20% fold change in expression.)
- How many genes have B&H corrected p < 0.05?
- How many genes have a Bonferroni corrected p < 0.05?
- Use this sample PowerPoint slide to see how your table should be formatted.
Between-strain ANOVA
The detailed description of how this is done can be found on this page. A brief version of the protocol appears below.
- All two strain comparisons were performed in MATLAB using the script Two_strain_compare_corrected_20140813_3pm.zip (within a zip file):
- Download the zipped script file, extract it to the folder that contains your Excel file with the worksheet named "Master_Sheet". (The script and Excel file must be in the same folder to work.)
- Launch MATLAB version 2014b.
- In MATLAB, you will need to navigate to the folder containing the script and the Excel file.
- Near the top of the page, you will see a a field that contains the path to the working directory. Just to the left of it, there is an icon that looks like a folder opening with a green down arrow. Click on this icon to open a dialog box where you can choose your folder containing the script and Excel file.
- Once you have selected your folder, the left-hand pane should display the contents of that folder. To open the MATLAB script, you can double-click on it from that pane. The code for the script will appear in the center pane.
- You will need to make a few edits to the code, depending on which strain comparison you want to make.
- For the first block of code, the user must input the name of the Excel file (
*.xls or .xlsx) to be imported as the variable "filename", the sheet from which the data will be imported as the variable "sheetname", and the two strains that will be compared as the variables "strain1" and "strain2".- MATLAB will read either .xls or .xlsx
- Also note that this script will not work for any comparison involving dSWI4 because it has been hard-coded to expect 5 timepoints instead of 4.
- For the first block of code, the user must input the name of the Excel file (
%% User must input filename, sheetname, and strains for comparison filename = 'GCAT_and_Ontario_Final_Normalized_Data.xls'; % Name of input file sheetname = 'Master_Sheet'; % Name of sheet in input file containing data to analyze % % If one of the two strains you are working on is the wildtype, keep that % % wildtype as strain 1. strain1 = 'wt'; %Here should be wt, dCIN5, dGLN3, dHAP4, dHMO1, dZAP1, or Spar % % Select strain 2 to be one of the other strains you would like to % % compare with the first strain. strain2 = 'dZAP1'; %Here should be wt, dCIN5, dGLN3, dHAP4, dHMO1, dZAP1, or Spar
- The user does not have to modify any of the code from here on.
- The next two lines of code ask the user whether or not they would like to see plots for each gene with an unadjusted p-value < 0.05. If the user does want to see these plots, they enter "1". If they would not like to see these plots, the user enters "0". When prompted, enter a "1" to see the plots displayed.
disp('Do you want to view plots for each gene with an unadjusted p-value < 0.05?')
graph = input('If yes, enter "1". If no, enter "0". ');
- Plots will show for all genes with p < 0.05, which could be hundreds. If you want, you can save individual plots from the graph window, they do not save automatically. Press any key to display the next plot. The script will not finish and output the results files until all of the plots have been viewed.
- Because MATLAB version 2014b has switched to the OpenGL graphics library, the plots look weird at this point.
Step 7-8: Clustering and GO Term Enrichment with stem
- Prepare your microarray data file for loading into STEM.
- Insert a new worksheet into your Excel workbook, and name it "(STRAIN)_stem".
- Select all of the data from your "(STRAIN)_ANOVA" worksheet and Paste special > paste values into your "(STRAIN)_stem" worksheet.
- Your leftmost column should have the column header "MasterIndex". Rename this column to "SPOT". Column B should be named "ID". Rename this column to "Gene Symbol". Delete the column named "StandardName".
- Filter the data on the B-H corrected p value to be > 0.05 (that's greater than in this case).
- Once the data has been filtered, select all of the rows (except for your header row) and delete the rows by right-clicking and choosing "Delete Row" from the context menu. Undo the filter. This ensures that we will cluster only the genes with a "significant" change in expression and not the noise.
- Delete all of the data columns EXCEPT for the Average Log Fold change columns for each timepoint (for example, wt_AvgLogFC_t15, etc.).
- Rename the data columns with just the time and units (for example, 15m, 30m, etc.).
- Save your work. Then use Save As to save this spreadsheet as Text (Tab-delimited) (*.txt). Click OK to the warnings and close your file.
- Note that you should turn on the file extensions if you have not already done so.
- Now download and extract the STEM software. Click here to go to the STEM web site.
- Click on the download link, register, and download the
stem.zipfile to your Desktop. - Unzip the file. In Seaver 120, you can right click on the file icon and select the menu item 7-zip > Extract Here.
- This will create a folder called
stem. Inside the folder, double-click on thestem.jarto launch the STEM program.
- Click on the download link, register, and download the
- Running STEM
- In section 1 (Expression Data Info) of the the main STEM interface window, click on the Browse... button to navigate to and select your file.
- Click on the radio button No normalization/add 0.
- Check the box next to Spot IDs included in the data file.
- In section 2 (Gene Info) of the main STEM interface window, select Saccharomyces cerevisiae (SGD), from the drop-down menu for Gene Annotation Source. Select No cross references, from the Cross Reference Source drop-down menu. Select No Gene Locations from the Gene Location Source drop-down menu.
- In section 3 (Options) of the main STEM interface window, make sure that the Clustering Method says "STEM Clustering Method" and do not change the defaults for Maximum Number of Model Profiles or Maximum Unit Change in Model Profiles between Time Points.
- In section 4 (Execute) click on the yellow Execute button to run STEM.
- In section 1 (Expression Data Info) of the the main STEM interface window, click on the Browse... button to navigate to and select your file.
- Viewing and Saving STEM Results
- A new window will open called "All STEM Profiles (1)". Each box corresponds to a model expression profile. Colored profiles have a statistically significant number of genes assigned; they are arranged in order from most to least significant p value. Profiles with the same color belong to the same cluster of profiles. The number in each box is simply an ID number for the profile.
- Click on the button that says "Interface Options...". At the bottom of the Interface Options window that appears below where it says "X-axis scale should be:", click on the radio button that says "Based on real time". Then close the Interface Options window.
- Take a screenshot of this window (on a PC, simultaneously press the
AltandPrintScreenbuttons to save the view in the active window to the clipboard) and paste it into a PowerPoint presentation to save your figures.
- Click on each of the SIGNIFICANT profiles (the colored ones) to open a window showing a more detailed plot containing all of the genes in that profile.
- Take a screenshot of each of the individual profile windows and save the images in your PowerPoint presentation.
- At the bottom of each profile window, there are two yellow buttons "Profile Gene Table" and "Profile GO Table". For each of the profiles, click on the "Profile Gene Table" button to see the list of genes belonging to the profile. In the window that appears, click on the "Save Table" button and save the file to your desktop. Make your filename descriptive of the contents, e.g. "wt_profile#_genelist.txt", where you replace the number symbol with the actual profile number.
- Upload these files to LionShare and e-mail a link to Dr. Dahlquist. (It will be easier to zip all the files together and upload them as one file).
- For each of the significant profiles, click on the "Profile GO Table" to see the list of Gene Ontology terms belonging to the profile. In the window that appears, click on the "Save Table" button and save the file to your desktop. Make your filename descriptive of the contents, e.g. "wt_profile#_GOlist.txt", where you use "wt", "dGLN3", etc. to indicate the dataset and where you replace the number symbol with the actual profile number. At this point you have saved all of the primary data from the STEM software and it's time to interpret the results!
- Upload these files to LionShare and e-mail a link to Dr. Dahlquist. (It will be easier to zip all the files together and upload them as one file).
- A new window will open called "All STEM Profiles (1)". Each box corresponds to a model expression profile. Colored profiles have a statistically significant number of genes assigned; they are arranged in order from most to least significant p value. Profiles with the same color belong to the same cluster of profiles. The number in each box is simply an ID number for the profile.
- Analyzing and Interpreting STEM Results
- Select one of the profiles you saved in the previous step for further intepretation of the data. I suggest that you choose one that has a pattern of up- or down-regulated genes at the early (first three) timepoints. You and your partner will choose the same profile so that you can compare your results between the two strains. Answer the following:
- Why did you select this profile? In other words, why was it interesting to you?
- How many genes belong to this profile?
- How many genes were expected to belong to this profile?
- What is the p value for the enrichment of genes in this profile? Bear in mind that we just finished computing p values to determine whether each individual gene had a significant change in gene expression at each time point. This p value determines whether the number of genes that show this particular expression profile across the time points is significantly more than expected.
- Open the GO list file you saved for this profile in Excel. This list shows all of the Gene Ontology terms that are associated with genes that fit this profile. Select the third row and then choose from the menu Data > Filter > Autofilter. Filter on the "p-value" column to show only GO terms that have a p value of < 0.05. How many GO terms are associated with this profile at p < 0.05? The GO list also has a column called "Corrected p-value". This correction is needed because the software has performed thousands of significance tests. Filter on the "Corrected p-value" column to show only GO terms that have a corrected p value of < 0.05. How many GO terms are associated with this profile with a corrected p value < 0.05?
- Select 10 Gene Ontology terms from your filtered list (either p < 0.05 or corrected p < 0.05).
- Since you and your partner are going to compare the results from each strain for the same cluster, you can either:
- Choose the same 10 terms that are in common between strains.
- Choose 10 terms that are different between the strains (5 or so from each).
- Choose some that are the same and some that are different.
- Look up the definitions for each of the terms at http://geneontology.org. For your final lab report, you will discuss the biological interpretation of these GO terms. In other words, why does the cell react to cold shock by changing the expression of genes associated with these GO terms? Also, what does this have to do with HAP4 being deleted?
- To easily look up the definitions, go to http://geneontology.org.
- Copy and paste the GO ID (e.g. GO:0044848) into the search field at the upper left of the page called "Search GO Data".
- In the results page, click on the button that says "Link to detailed information about <term>, in this case "biological phase"".
- The definition will be on the next results page, e.g. here.
- Since you and your partner are going to compare the results from each strain for the same cluster, you can either:
- Select one of the profiles you saved in the previous step for further intepretation of the data. I suggest that you choose one that has a pattern of up- or down-regulated genes at the early (first three) timepoints. You and your partner will choose the same profile so that you can compare your results between the two strains. Answer the following:
Step 9: GenMAPP & MAPPFinder
Preparing the Input File for GenMAPP
- Insert a new worksheet and name it STRAIN_GenMAPP.
- Go back to the "ANOVA" worksheet for your strain and Select All and Copy.
- Go to your new sheet and click on cell A1 and select Paste Special, click on the Values radio button, and click OK.
- Delete the columns containing the "ss" calculations, just retaining the individual log fold change data, the average log fold change data, and the p values. For the Bonferroni and B&H p values, just keep the one column where we replaced all values > 1 with 1.
- Now go to your "_ttest" worksheet. Copy just the columns containing the P values for the individual timepoints and Paste special > Paste values into your GenMAPP worksheet to the right of the previous data. For the Bonferroni and B&H p values, just keep one column where we replaced all values > 1 with 1.
- It will be useful if we arrange the columns in a slightly different order: all individual log fold change data, then the ANOVA p values, then the AvgLogFC and p values for the individual timepoints clustered together (e.g., all t15 data together).
- Go to the Excel file that was produced by MATLAB with the between-strain ANOVA results.
- Copy and paste Column S (p value) and Column V into the next columns to the right of your GenMAPP worksheet. Rename the columns "wt_v_STRAIN_Pval" and "wt_v_STRAIN_B-H-Pval".
- Select all of the columns containing Fold Changes. Select the menu item Format > Cells. Under the number tab, select 2 decimal places. Click OK.
- Select all of the columns containing P values. Select the menu item Format > Cells. Under the number tab, select 4 decimal places. Click OK.
- We will now format this file for use with GenMAPP.
- Currently, the "MasterIndex" column is the first column in the worksheet. We need the "ID" column to be the first column. Select Column B and Cut. Right-click on Cell A1 and select "Insert cut cells". This will reverse the position of the columns.
- Insert a new empty column in Column B. Type "SystemCode" in the first cell and "D" in the second cell of this column. Use our trick to fill this entire column with "D".
- Make sure to save this work as your .xlsx file. Now save this worksheet as a tab-delimited text file for use with GenMAPP in the next section.
Running GenMAPP
Each time you launch GenMAPP, you need to make sure that the correct Gene Database (.gdb) is loaded.
- Look in the lower left-hand corner of the window to see which Gene Database has been selected.
- If you need to change the Gene Database, select Data > Choose Gene Database. Navigate to the directory C:\GenMAPP 2 Data\Gene Databases and choose the correct one for your species.
- For the exercise today, if the yeast Gene Database is not present on your computer, you will need to download it. Click this link to download the yeast Gene Database.
- Unzip the file and save it, Sc-Std_20060526.gdb, to the folder C:\GenMAPP 2 Data\Gene Databases.
GenMAPP Expression Dataset Manager Procedure
- Launch the GenMAPP Program. Check to make sure the correct Gene Database is loaded.
- Select the Data menu from the main Drafting Board window and choose Expression Dataset Manager from the drop-down list. The Expression Dataset Manager window will open.
- Select New Dataset from the Expression Datasets menu. Select the tab-delimited text file that you formatted for GenMAPP (.txt) in the procedure above from the file dialog box that appears.
- The Data Type Specification window will appear. GenMAPP is expecting that you are providing numerical data. If any of your columns has text (character) data, check the box next to the field (column) name.
- The column StandardName has text data in it, but none of the rest do.
- Allow the Expression Dataset Manager to convert your data.
- This may take a few minutes depending on the size of the dataset and the computer’s memory and processor speed. When the process is complete, the converted dataset will be active in the Expression Dataset Manager window and the file will be saved in the same folder the raw data file was in, named the same except with a .gex extension; for example, MyExperiment.gex.
- A message may appear saying that the Expression Dataset Manager could not convert one or more lines of data. Lines that generate an error during the conversion of a raw data file are not added to the Expression Dataset. Instead, an exception file is created. The exception file is given the same name as your raw data file with .EX before the extension (e.g., MyExperiment.EX.txt). The exception file will contain all of your raw data, with the addition of a column named ~Error~. This column contains either error messages or, if the program finds no errors, a single space character.
- I had 97 errors
- Customize the new Expression Dataset by creating new Color Sets which contain the instructions to GenMAPP for displaying data on MAPPs.
- Color Sets contain the instructions to GenMAPP for displaying data from an Expression Dataset on MAPPs. Create a Color Set by filling in the following different fields in the Color Set area of the Expression Dataset Manager: a name for the Color Set, the gene value, and the criteria that determine how a gene object is colored on the MAPP. Enter a name in the Color Set Name field that is 20 characters or fewer. You will have one Color Set per strain per time point.
- The Gene Value is the data displayed next to the gene box on a MAPP. Select the column of data to be used as the Gene Value from the drop down list or select [none]. We will use "Avg_LogFC_" for the the appropriate time point.
- Activate the Criteria Builder by clicking the New button.
- Enter a name for the criterion in the Label in Legend field.
- Choose a color for the criterion by left-clicking on the Color box. Choose a color from the Color window that appears and click OK.
- State the criterion for color-coding a gene in the Criterion field.
- A criterion is stated with relationships such as "this column greater than this value" or "that column less than or equal to that value". Individual relationships can be combined using as many ANDs and ORs as needed. A typical relationship is
[ColumnName] RelationalOperator Value
with the column name always enclosed in brackets and character values enclosed in single quotes. For example:
[Fold Change] >= 2 [p value] < 0.05 [Quality] = 'high'
This is the equivalent to queries that you performed on the command line when working with the PostgreSQL movie database. GenMAPP is using a graphical user interface (GUI) to help the user format the queries correctly. The easiest and safest way to create criteria is by choosing items from the Columns and Ops (operators) lists shown in the Criteria Builder. The Columns list contains all of the column headings from your Expression Dataset. To choose a column from the list, click on the column heading. It will appear at the location of the cursor in the Criterion box. The Criteria Builder surrounds the column names with brackets.
The Ops (operators) list contains the relational operators that may be used in the criteria: equals ( = ) greater than ( > ), less than ( < ), greater than or equal to ( >= ), less than or equal to ( <= ), is not equal to ( <> ). To choose an operator from the list, click on the symbol. It will appear at the location of the insertion bar (cursor) in the Criterion box. The Criteria Builder automatically surrounds the operators with spaces. The Ops list also contains the conjunctions AND and OR, which may be used to make compound criteria. For example:
[Fold Change] > 1.2 AND [p value] <= 0.05
Parentheses control the order of evaluation. Anything in parentheses is evaluated first. Parentheses may be nested. For example:
[Control Average] = 100 AND ([Exp1 Average] > 100 OR [Exp2 Average] > 100)
Column names may be used anywhere a value can, for example:
[Control Average] < [Experiment Average]
- After completing a new criterion, add the criterion entry (label, criterion, and color) to the Criteria List by clicking the Add button.
- For the yeast dataset, you will create two criterion for each Color Set. "Increased" will be [<strain>_Avg_LogFC_<timepoint>] > 0.25 AND [<strain>Pval_<timepoint>] < 0.05 and "Decreased will be [<strain>_Avg_LogFC_<timepoint>] < -0.25 AND [<strain>Pval_<timepoint>] < 0.05. Make sure that the increased and decreased average log fold change values match the timepoint of the Color Set.
- You may continue to add criteria to the Color Set by using the previous steps.
- The buttons to the right of the list represent actions that can be performed on individual criteria. To modify a criterion label, color, or the criterion itself, first select the criterion in the list by left-clicking on it, and then click the Edit button. This puts the selected criterion into the Criteria Builder to be modified. Click the Save button to save changes to the modified criterion; click the Add button to add it to the list as a separate criterion. To remove a criterion from the list, left-click on the criterion to select it, and then click on the Delete button. The order of Criteria in the list has significance to GenMAPP. When applying an Expression Dataset and Color Set to a MAPP, GenMAPP examines the expression data for a particular gene object and applies the color for the first criterion in the list that is true. Therefore, it is imperative that when criteria overlap the user put the most important or least inclusive criteria in the list first. To change the order of the criteria in the list, left-click on the criterion to select it and then click the Move Up or Move Down buttons. No criteria met and Not found are always the last two positions in the list.
- You will also create a ColorSets to view the within-strain ANOVA p values for your strain, with criteria for viewing the unadjusted, Bonferroni-corrected, and B&H corrected p values.
- Finally, you will create a ColorSet to view the between-strain ANOVA p values for your wt v. STRAIN comparison.
- Save the entire Expression Dataset by selecting Save from the Expression Dataset menu. Changes made to a Color Set are not saved until you do this.
- Exit the Expression Dataset Manager to view the Color Sets on a MAPP. Choose Exit from the Expression Dataset menu or click the close box in the upper right hand corner of the window.
- Upload your .gex file to Lionshare and share it with Dr. Dahlquist. E-mail the link to the file to Dr. Dahlquist.
- Click here to download a zipped set of MAPPs with which to view your Expression Dataset.
Step 10: YEASTRACT
Using YEASTRACT to Infer which Transcription Factors Regulate a Cluster of Genes
In the previous analysis using STEM, we found a number of gene expression profiles (aka clusters) which grouped genes based on similarity of gene expression changes over time. The implication is that these genes share the same expression pattern because they are regulated by the same (or the same set) of transcription factors. We will explore this using the YEASTRACT database.
- Open the gene list in Excel for the one of the significant profiles from your stem analysis. Choose a cluster with a clear cold shock/recovery up/down or down/up pattern. You should also choose one of the largest clusters.
- Copy the list of gene IDs onto your clipboard.
- Launch a web browser and go to the YEASTRACT database.
- On the left panel of the window, click on the link to Rank by TF.
- Paste your list of genes from your cluster into the box labeled ORFs/Genes.
- Check the box for Check for all TFs.
- Accept the defaults for the Regulations Filter (Documented, DNA binding plus expression evidence)
- Do not apply a filter for "Filter Documented Regulations by environmental condition".
- Rank genes by TF using: The % of genes in the list and in YEASTRACT regulated by each TF.
- Click the Search button.
- Answer the following questions:
- In the results window that appears, the p values colored green are considered "significant", the ones colored yellow are considered "borderline significant" and the ones colored pink are considered "not significant". How many transcription factors are green or "significant"?
- List the "significant" transcription factors on your wiki page, along with the corresponding "% in user set", "% in YEASTRACT", and "p value".
- Are CIN5, GLN3, HAP4, HMO1, SWI4, and ZAP1 on the list?
- For the mathematical model that we will build, we need to define a gene regulatory network of transcription factors that regulate other transcription factors. We can use YEASTRACT to assist us with creating the network. We want to generate a network with approximately 15-30 transcription factors in it.
- You need to select from this list of "significant" transcription factors, which ones you will use to run the model. You will use these transcription factors and add CIN5, GLN3, HAP4, HMO1, SWI4, and ZAP1 if they are not in your list. Explain in your electronic notebook how you decided on which transcription factors to include. Record the list and your justification in your electronic lab notebook.
- Go back to the YEASTRACT database and follow the link to Generate Regulation Matrix.
- Copy and paste the list of transcription factors you identified (plus CIN5, HAP4, GLN3, HMO1, SWI4, and ZAP1) into both the "Transcription factors" field and the "Target ORF/Genes" field.
- We are going to generate several regulation matrices, with different "Regulations Filter" options.
- For the first one, accept the defaults: "Documented", "DNA binding plus expression evidence"
- Click the "Generate" button.
- In the results window that appears, click on the link to the "Regulation matrix (Semicolon Separated Values (CSV) file)" that appears and save it to your Desktop. Rename this file with a meaningful name so that you can distinguish it from the other files you will generate.
- Repeat these steps to generate a second regulation matrix, this time applying the Regulations Filter "Documented", "Only DNA binding evidence".
- Repeat these steps a third time to generate a third regulation matrix, this time applying the Regulations Filter "Documented", DNA binding and expression evidence".
Visualizing Your Gene Regulatory Networks with GRNsight
We will analyze the regulatory matrix files you generated above in Microsoft Excel and visualize them using GRNsight to determine which one will be appropriate to pursue further in the modeling.
- First we need to properly format the output files from YEASTRACT. You will repeat these steps for each of the three files you generated above.
- Open the file in Excel. It will not open properly in Excel because a semicolon was used as the column delimiter instead of a comma. To fix this, Select the entire Column A. Then go to the "Data" tab and select "Text to columns". In the Wizard that appears, select "Delimited" and click "Next". In the next window, select "Semicolon", and click "Next". In the next window, leave the data format at "General", and click "Finish". This should now look like a table with the names of the transcription factors across the top and down the first column and all of the zeros and ones distributed throughout the rows and columns. This is called an "adjacency matrix." If there is a "1" in the cell, that means there is a connection between the trancription factor in that row with that column.
- Save this file in Microsoft Excel workbook format (.xlsx).
- Check to see that all of the transcription factors in the matrix are connected to at least one of the other transcription factors by making sure that there is at least one "1" in a row or column for that transcription factor. If a factor is not connected to any other factor, delete its row and column from the matrix. Make sure that you still have somewhere between 15 and 30 transcription factors in your network after this pruning.
- Only delete the transcription factor if there are all zeros in its column AND all zeros in its row. You may find visualizing the matrix in GRNsight (below) can help you find these easily.
- For this adjacency matrix to be usable in GRNmap (the modeling software) and GRNsight (the visualization software), we need to transpose the matrix. Insert a new worksheet into your Excel file and name it "network". Go back to the previous sheet and select the entire matrix and copy it. Go to you new worksheet and click on the A1 cell in the upper left. Select "Paste special" from the "Home" tab. In the window that appears, check the box for "Transpose". This will paste your data with the columns transposed to rows and vice versa. This is necessary because we want the transcription factors that are the "regulatORS" across the top and the "regulatEES" along the side.
- The labels for the genes in the columns and rows need to match. Thus, delete the "p" from each of the gene names in the columns. Adjust the case of the labels to make them all upper case.
- In cell A1, copy and paste the text "rows genes affected/cols genes controlling".
- Now we will visualize what these gene regulatory networks look like with the GRNsight software.
- Go to the GRNsight home page (you can either use the version on the home page or the beta version.
- Select the menu item File > Open and select one of the regulation matrix .xlsx file that has the "network" worksheet in it that you formatted above. If the file has been formatted properly, GRNsight should automatically create a graph of your network. Move the nodes (genes) around until you get a layout that you like and take a screenshot of the results. Paste it into your PowerPoint presentation. Repeat with the other two regulation matrix files. You will want to arrange the genes in the same order for each screenshot so that the graphs can be easily compared.
Step 11: GRNmap
Create the Input Excel Workbook for the Model
- Your file will be similar to the file "21-genes_50-edges_Dahlquist-data_Sigmoid_estimation.xls", but with your expression data and network. You should download this file, change the name, and edit it to include your data. Make sure to give it a meaningful filename that includes your last name or initials. Click this link to download the sample file from the GRNmap GitHub repository.)
- The first thing you need to do is determine the transcription factors that you are including in your network. You are going to use the "transposed" Regulation Matrix that you generated from YEASTRACT in the previous section.
- Copy the transposed matrix from your "network" sheet and paste it into the worksheets called "network" and "network_weights".
- Note that the transcription factor names have to be in the same order and same format across the top row and first column. CIN5 does not match Cin5p, so the latter will need to be changed to CIN5 if you have not already done so.
- It may be easier for you if you put the transcription factors in alphabetical order (using the sort feature in Excel), but whether you leave your list the same as it is from the YEASTRACT assignment or in alphabetical order, make sure it is the same order for all of the worksheets.
- The next worksheet to edit is the one called "degradation_rates".
- Paste your list of transcription factors from your "network" sheet into the column named "id".
- Next, you will need to look up the degradation rates for your list of transcription factors. These rates have been calculated from protein half-life data from a paper by Belle et al. (2006). Look up the rates for your transcription factors from this file and include them in your "degradation_rates" worksheet.
- If a transcription factor does not appear in the file above, use the value "0.027182242" for the degradation rate.
- The next worksheet to edit is the one called "production_rates".
- Paste the "id" column from your "degradation_rates" sheet into the "production_rates" sheet.
- The initial guesses for the production rates we are using for the model are two times the degradation rate. Compute these values from your degradation rates and paste the values into the column titled "production_rate".
- Next you will input the expression data for the wild type strain and one other strain (dcin5, dgln3, dhap4, dhmo1, dzap1, or spar; note that we can't use dswi4 because it only has 2 cold shock timepoints). You need to include only the data for the genes in your network, in the same order as they appear in the other worksheets.
- Put the wild type data in the sheet called "wt".
- The sample spreadsheet has a worksheet named "dcin5". Change this name to match the strain you are using (listed above). The instructions below should be followed for each strain sheet.
- Paste the "id" column from one of your previous sheets into this one.
- This data in this sheet is the Log Fold Changes for each replicate and each timepoint from the "Rounded_Normalized_Data" worksheet from the big Excel workbook in which you computed the statistics. We are only going to use the cold shock timepoints for the modeling. Thus your column headings for the data should be "15", "30", and "60". There will be multiple columns for each timepoint (typically 4) to represent the replicate data, but they will all have the same name. For example, you may have four columns with the header "15".
- Copy and paste the data from your spreadsheet into this one. You need to include only the data for the genes in your network. Make sure that the genes are in the same order as in the other sheets.
- The "optimization_parameters" worksheet should have the following values:
- alpha should be 0.002
- kk_max should be 1
- MaxIter should be 1e08 (one hundred million in plain English)
- TolFun should be 1e-6
- MaxFunEval should be 1e08 (one hundred million in plain English)
- TolX should be 1e-6
- production_function should be Sigmoid (case-insensitive)
- estimate_params should be 1
- make_graphs should be 1
- fix_P should be 0
- fix_b should be 0
- For the parameter "expression_timepoints" (Cell A13), we should have "15", "30", and "60", since these are the timepoints we have in our data.
- For the parameter "Strain" (Cell A14), replace "dcin5" with the name of the second strain you are using, making sure that the capitalizaiton and spelling is the same as what you named the worksheet containing that strain's expression data. We are only going to compare two strains, so you can delete the other strain information.
- For the parameter "simulation_timepoints" (Cell A15), perform the forward simulation of the expression in five minute increments from 0 to 60 minutes. Thus, this row should read: simtime should be 0, 5, <...fill by steps of 5...>, 60, each number in a different cell.
- The last sheet you will need to modify is called "threshold_b".
- Paste in the list of standard names for your transcription factors from one of your previous sheets.
- Cell A1 in the sample files has the text "rows genes affected/cols genes controlling". I believe you can either have this text in cell A1 or "id".
- The "threshold" value for each gene should be "0".
- When you have completed the modifications to your file, upload it to LionShare and send Dr. Dahlquist an e-mail with a link to the file.
Appendix: Full explanation of the "optimization_parameters" sheet
alpha: Penalty term weighting (from an L-curve analysis)kk_max: Number of times to re-run the optimization loop: in some cases re-starting the optimization loop can improve performance of the estimation.MaxIter: Number of times MATLAB iterates through the optimization scheme. If this is set too low, MATLAB will stop before the parameters are optimized.TolFun: How different two least squares evaluations should be before it says it's not making any improvementMaxFunEval: maximum number of times it will evaluate the least squares costTolX: How close successive least squares cost evaluations should be before MATLAB determines that it is not making any improvement.production_function:= Sigmoid(case-insensitive) if sigmoidal model,=MM(case-insensitive) if Michaelis-Menten modelL_curve:=0if an L-curve analysis should NOT be run or=1if an L-curve analysis SHOULD be run.- The L-curve analysis will automatically run sequential rounds of estimation for an array of fixed alpha values (0.8, 0.5, 0.2, 0.1,0.08, 0.05,0.02,0.01, 0.008, 0.005, 0.002, 0.001, 0.0008, 0.0005, 0.0002, and 0.0001). GRNmap makes a copy of the user's selected input workbook and changes alpha to the first alpha in the list. The estimation runs and the resulting parameter values are used as the initial guesses for the next round of estimation with the next alpha value. This process repeats until all alpha values have been run. New input and output workbooks are generated for each alpha value, although currently, the graphs are only saved for the last run.
estimate_params:=1if want to estimate parameters and=0if the user wants to do just one forward runmake_graphs:=1to output graphs;=0to not output graphsfix_P:=1if the user does not want to estimate the production rate, P, parameter, use initial guess and never change;=0to estimatefix_b:=1if the user does not want to estimate the b parameter, use initial guess and never change;=0to estimateexpression_timepoints: A row containing a list of the time points when the data was collected experimentally. Should correspond to the timepoint column headers in the expression sheets.Strain: A row containing a list of all of the strains for which there is expression data in the workbook. Should correspond to the names of the sheets for each strain.simulation_timepoints: A list of times for which the forward simulation should be evaluated.
Running GRNmap
You will now finally run the GRNmap model on the input workbook you created above. You will run the optimization twice; once where the threshold parameters, b, are not estimated and once where the threshold parameters 'are estimated. You will compare the estimated weight and production rate parameters outputted by these two runs with each other.
- Download the current version of GRNmap from GitHub. Version 1.0.6 can be downloaded by following this link.
- For the sake of organization, save it into a new folder called "GRNmap" either on your Desktop or within your "Microarray Analysis" folder.
- Unzip the file by right-clicking on it and choosing 7-zip > Extract here.
- Open the "GRNmap-1.0.6" folder and open the "matlab" subfolder. Double-click on the file "GRNmodel.m" to open GRNmap in MATLAB 2014b.
- Click on the green triangle "Run" button to run the model.
- You will be prompted by an Open dialog to find your input file that you created in the previous section. Browse and select this input file and click OK.
- Note that the Open dialog will default to show files of
*.xlsxonly. If your file is saved as*.xls, you will need to select the drop-down menu to show all files. - A window called "Figure 1" will appear. The counter is showing the number of iterations of the least squares optimization algorithm. The top plot is showing the values of all the parameters being estimated. You should see some movement of the diamonds each time the counter iterates.
- Once the model has completed its run, plots showing the expression over time for all of the genes in the network will appear. Since we selected "makeGraphs = 1" these will automatically be saved as
*.jpgfiles in the same folder as your input file. Compile the figures into a single PowerPoint file. Please label things clearly, placing an appropriate number of graphs on each page for a readable visual. Take some care to make sure that the graphs are the same size and the aspect ratio has not been changed. - Create a new workbook for analyzing the weight data. In this workbook, create a new sheet: call it estimated_weights. In this new worksheet, create a column of labels of the form ControllerGeneA -> TargetGeneB, replacing these generic names with the standard gene names for each regulatory pair in your network. Remember that columns represent Controllers and rows represent Targets in your network and network_weights sheets.
- Extract the non-zero optimized weights from their worksheet and put them in a single column next to the corresponding ControllerGeneA -> TargetGeneB label.
- Now we will run the model a second time, this time estimating the threshold parameters, b. Save the input workbook that you previously created as a new file with a meaningful name (e.g. append "estimate-b" to the previous filename), and change fix_b to 0 in the "optimization_parameters" worksheet, so that the thresholds will be estimated. Rerun GRNmodel with the new input sheet.
- Repeat Parts (4) through (6) with the new output.
- Create an empty excel workbook, and copy both sets of weights into a worksheet.
- Create a bar chart in order to compare the "fixed b" and "estimated b" weights.
- Create bar charts to compare the production rates from each run.
- Copy the two bar charts into your powerpoint.
- Visualize the output of each of your model runs with GRNsight.
- In order for this to work, you need to alter your output workbook slightly. You need to change the name of the sheet called "out_network_optimized_weights" to "network_optimized_weights"; i.e., delete the "out_" from that sheet name.
- Arrange the genes in the same order you used to display them previously when you visualized the networks from YEASTRACT for both of your model output runs. Take a screenshot of each of the results and paste it into your PowerPoint presentation. Clearly label which screenshot belongs to which run.
- Note that GRNsight will display differently now that you have estimated the weights. For positive weights > 0, the edge will be given a regular (pointy) arrowhead to indicate an activation relationship between the two nodes. For negative weights < 0, the edge will be given a blunt arrowhead (a line segment perpendicular to the edge direction) to indicate a repression relationship between the two nodes. The thickness of the edge will vary based on the magnitude of the absolute value of the weight. Larger magnitudes will have thicker edges and smaller magnitudes will have thinner edges. The way that GRNsight determines the edge thickness is as follows. GRNsight divides all weight values by the absolute value of the maximum weight in the matrix to normalize all the values to between zero and 1. GRNsight then adjusts the thickness of the lines to vary continuously from the minimum thickness (for normalized weights near zero) to maximum thickness (normalized weights of 1). The color of the edge also imparts information about the regulatory relationship. Edges with positive normalized weight values from 0.05 to 1 are colored magenta; edges with negative normalized weight values from -0.05 to -1 are colored cyan. Edges with normalized weight values between -0.05 and 0.05 are colored grey to emphasize that their normalized magnitude is near zero and that they have a weak influence on the target gene.
- Upload your PowerPoint, your two input workbooks, and your two output workbooks and link to them in your individual journal. Also upload the workbook where you made the bar charts comparing the weights from both runs.
- Interpret the results of the model simulation.
- Examine the graphs that were output by each of the runs. Which genes in the model have the closest fit between the model data and actual data? Which genes have the worst fit between the model and actual data? Why do you think that is? (Hint: how many inputs do these genes have?) How does this help you to interpret the microarray data?
- Which genes showed the largest dynamics over the timecourse? In other words, which genes had a log fold change that is different than zero at one or more timepoints. The p values from the Week 11 ANOVA analysis are informative here. Does this seem to have an effect on the goodness of fit (see question above)?
- Which genes showed differences in dynamics between the wild type and the other strain your group is using? Does the model adequately capture these differences? Given the connections in your network (see the visualization in GRNsight), does this make sense? Why or why not?
- Examine the bar charts comparing the weights and production rates between the two runs. Were there any major differences between the two runs? Why do you think that was? Given the connections in your network (see the visualization in GRNsight), does this make sense? Why or why not?
- Finally, based on the results of your entire project, which transcription factors are most likely to regulate the cold shock response and why?
- Based on these results, what future directions do you want to take?
- Interpret the results of the model simulation.
