Cre-Lox
Overall Idea
- PCR Gene of Interest
- Clean up PCR / Purify Plasmid
- Restriction Digest (create sticky ends)
- Low melt gel to purify product from fragments. Slice gel products
- Ligation melt - melt sliced gel products, combine plasmid gel slice with PCR gel slice, add ligase buffer + BSA
- Transform new plasmid
- Colony PCR to check if previous steps worked
- Purify plasmid from colonies that worked
REPEAT to insert additional genes
Protocols
February
2/28/2012
1.PCR product!
PCR
10 ul Hf (5x) buffer
1.5 ul dNTP
1 ul plasmid (181 or 125)
1 ul forward primer
1 ul reverse primer
35 ul H2O
.5 ul Phusion polymerase
50 ul total
March
3/8/2012
1.Cre PCR? It hasn’t worked on the gel yet
a.Dr. Grose said to reevaluate the cre primer and see what must be going wrong with it.
2.Restriction digest with Lox
a.Eric did the PCR DNA purification for the Lox so we need to start with the restriction digest today
b.We did a restriction enzyme with both the PCR and the plasmid (PIG12) when we did the restriction digest with the plasmid there was a question if enough EcoRI made it into the aliquot. It was almost gone, so if there are any problems it could be linked to the lack of EcoRI in the plasmid digest.
c.What is the process for restriction digest:
41.4 ul PCR
5 ul buffer 4
.5 ul BSA
(mix all of those well before adding the enzymes)
1.5 ul IG11 pLat Forward
1.5 ul IG12 pLat Reverse
50ul total
37 degrees about 2 hours
Plasmid: (Digest plasmid only)
20 ul plasmid
21.5 ul ddH20
5 ul buffer 4
.5 ul BSA
(mix well before adding the enzymes)
1.5 Sac I
1.5 EcoRI-HF (high fidelity)
50 ul total
3/10/2012
Then we ran both restriction digests on the gel:
PCR Product for lox- 7th spot
Plasmid – 8th spot
Results:
3/13/2012
1.We need to locate the bacteria with the cre gene in it- this way we can purify the plasmid and start the PCR process over again since it has not worked thus far
We set up 2 reactions: vector+insert and a vector-only control.
6.5 μL ddH2O
1.5 μL 10X ligase buffer
3 μL vector
1 μL T4 DNA ligase
For vector+insert: 3 μL Lox insert
For vector-only: 3 μL ddH2O
3/15/2012
1. Cre- Dr. Grose located the template from Dr. Griffitts lab (PJG577). It is freshly plated so we will incubate it over night (37 degrees) and it should be ready to purify tomorrow to get the Cre PCR going again.
2. Lox- We used our lox ligation and transformed it. Here is the transformation protocol we used:
thaw competent cells on ice for ~20 min
add 2ul of ligation to ~25ul of competent cells - vortex or flick
put back on ice for 5-10 minutes
Heat shock at 42 degrees for 60 seconds and place immediately back on ice for 2-5 minutes
add .5 ml (500ul) of lb and incubate at 37 degrees (incubate 30 min if ampicillin and 60 min if anything else)
Plate 100ul of cells and incubate 37 degrees overnight
3/16/2012
Now that our Lox-transformed bacteria had been plated and grown, we chose 8 colonies from the plates. We test them to ensure that they have the transformed plasmid. We do this by boiling them to make template DNA, and then PCR-ing the multiple cloning site in order to see whether the gene we inserted made it in. The MCS is normally ~130 BP long, so after it is transformed, it ought to be 130BP+however many BP LOX is.
1. Added 1 colony to a PCR tube with 50 μL water.
2. Repeated 8 times. Carefully labelled the plate and the tube with numbers 1-8.
3. Set up the following TAQ PCR
19 μL H2O
2.5 μL Standard 10X reaction buffer
0.5 μL 10 mM dNTP's
0.5 μL each primer
0.5 μL TAQ DNA polymerase
Then added 2 μL of the boiled Lox-transformed samples.
3/20/2012
LOX: Today we ran a gel from the LOX colonies which were transformed. It ought to be 130BP+however many BP LOX is which will indicate that the insert is present. If the insert is not present then the results of the purified/opened up DNA will only show 130bp which is the size of only the multiple cloning site. The first column after the ladder is the colony that seems to have taken up the lox insert because it is 130BP+.
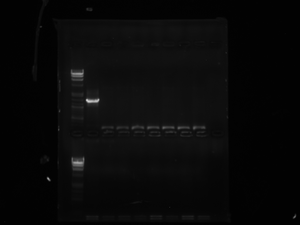
CRE: Since we grew the cre we streaked out from Dr. Griffitts lab last week we purified - we were able to use that as a template today to run our PCR which we will need to run on a gel Thursday 3/22/2012.
3/22/2012
Cre was run out on the following lanes: look in audrey's notebook

3/27/2012
CRE: The gel above shows that our Cre PCR did not work - we need to discuss with Dr. Grose why our PCR is still disfunctional. We are not sure where the template came from that Dr. Grose gave us.
LOX: Since or colony PCR had very low efficiency (1:8 colonies actually worked) we decided to run the colony PCR again with the same transformation to see if we could get any better results. We are going to run the LOX PCR today.
Now that our Lox-transformed bacteria had been plated and grown, we chose 8 colonies from the plates. We test them to ensure that they have the transformed plasmid. We do this by boiling them to make template DNA, and then PCR-ing the multiple cloning site in order to see whether the gene we inserted made it in. The MCS is normally ~130 BP long, so after it is transformed, it ought to be 130BP+however many BP LOX is.
1. Added 1 colony to a PCR tube with 50 μL water.
2. Repeated 8 times. Carefully labelled the plate and the tube with numbers 1-8.
3. Set up the following TAQ PCR
19 μL H2O
2.5 μL Standard 10X reaction buffer
0.5 μL 10 mM dNTP's
0.5 μL IG57 primer
0.5 μL pLAT reverse primer
0.5 μL TAQ DNA polymerase
Then added 2 μL of the boiled Lox-transformed samples.
3/29/2012
Lox gel: today we ran a gel with the new 8 colonies we were testing for the transformed plasmid. It doesn't look like any of the colonies worked. *Ours are the first 8 wells after the ladder on the bottom row. The efficency is still very low, something probably went bad with the restriction digest. We took the FIRST colony that worked from the FIRST gel we ran that worked and are now isolating the plasmid in the overnight at 37 degrees and we need to purify it tomorrow. (they are labeled BLANK and BLANK 1-1. The Blank 1-1 should be foggy and has the plasmid in it). They have the date 3.22.12 labelled on them.
Cre: We talked to Dr. Grose today about the Cre gene and she is going to get the exact sequence from Dr. Griffitts lab so we can triple check exactly what is not functioning. That is something we need to take care of ASAP.
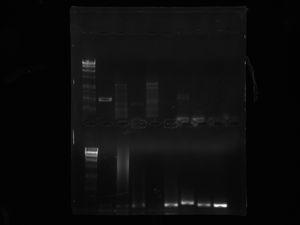
3/29/2012
Pelleted the bacteria with the lox and froze it in the Colon Cancer box.
April
4/28/2012 (HE and EJ)
Lox: today we did Plasmid miniprep kit. LABELLED PURIFIED LOX PLASMID. (it is in the e.coli box in the freezer)
- Harvest cells: Pellet 1-5 ml of an overnight recombinant E. Coli culture by centrifugation. (12,000xg for 1 min)
- Resuspend cells: completely resuspend the bacterial pellet with 200μL of the lysis solution. Immediately mix the contents by gentle inversion (6-8 times) DO NOT VORTEX! Do not allow lysis reaction to exceed 5 minutes.
- Neutralize: Add 350μL of the neutralization/binding solution. Gently invert the tube 4-6 times. Centrifuge 12,000xg max speed for 10min.
- Prepare column: Insert column into microcertrifuge tube. add 500ul of the column preparation solution to each miniprep column and centrifuge at 12,000xg for 30sec to 1min. discard the flow-through fluid.
- Load cleared lysate: transfer the cleared lysate from step 3 to the column prepared in step 4 and centrifuge for 30 sec to 1 min
- Optional wash (use only with EndA+ strains): Add 500ul of the optional wash solution to the column. Centrifuge for 30sec-1min. Discard flow through fluid.
- Wash column: Add 750ul of the diluted wash solution to the column. centrifuge for 30sec-1min. Discard flow through liquid and centrifuge again 1-2 min without any additional wash solution to remove excess ethanol.
- Elute DNA: transfer column to a fresh collection tube. Add 100ul of Elution Solution. Centrifuge 1min. The DNA is now present in the eluate and is ready for immediate storage at -20°C
Cre: we are still currently trying to attain the actual sequence for the PJG577 Cre gene from Dr. Griffitts lab to check our primers.
4/30/2012 (EJ and HE)
Lox: We need to determine what the construct of lox is so that we can have it sequenced by adding:
2ul plasmid
1ul primer
Cre: Dr. Griffitts lab is still looking for the cre sequence. They have not been able to find the sequence for the PJG577 and so they are going to continue to look for that and email it to us when they find it. Until then we are unable to make any progress with Cre
May
5/1/2012 EJ
Dr. Grose spoke with the Griffitts lab and discovered that the Cre sequence they use is different from the sequence we found on NCBI. That is why our primers didn't work.
This is the sequence for Cre from the Griffitts lab in pJG125.
Dr. Grose ordered new primers for this Cre sequence. They are BI155 and BI156. Dr. Grose also re-streaked the bacteria that contain the plasmid with Cre so we will have new template when the primers arrive. We can begin cloning this new Cre sequence as soon as we get the primers.
We need to figure out whether our lox plasmid has pBAD in it or not. Apparently some groups in the class used pLAT with pBAD and the other half used pLAT without pBAD. I don't really know how to figure out which one we used...
I (Eric) spoke with Julie and she thinks that we all used pIG12 which has the pBAD promoter in it.
Prepared the Lox plasmid to be sequenced:
In a PCR tube labeled Lox Plasmid Forward
2μL lox transformed plasmid
1μL BI12 (forward primer for pIG12)
In a second PCR tube labeled Lox Plasmid Reverse
2μL lox transformed plasmid
1μL BI157 (reverse primer for pIG12, to be renamed)
Gave the tubes to Dr. Grose who gave them to Jordan. Cross your fingers and hope everything worked!!!
5/4/2012 The best day so far.
HE and EJ
Cre: boiled the cre template (PJG125) from Dr. Griffitts lab so we are ready to start cloning the second the new cre primers arrive. We put it in the freezer in the Colon Cancer box labelled "Cre template."
Lox: We got the lox plasmid sequence back from the sequencing lab. We checked for similarities between the sequence and the lox term lox sequence and found a good match (E=0). So we are confident that lox got cloned in!!!!!!!!! Hurray!!!!!!!!!!!!
5/7/2012 The day after the best day so far. HE EJ KN
PCR amplify Cre gene
10 ul Hf (5x) buffer
1.5 ul 10mM dNTP
1 ul plasmid (pJG125 which was boiled last time by Hilary)
1 ul B155 (forward primer)
1 ul B156 (reverse primer)
35 ul ddH2OH2O
.5 ul Phusion polymerase
50 ul total
We wrote C2 on the top of the PCR tub and Cre PCR on the side.
This protocol will amplify the Cre gene from Griffitts lab from the plasmid pJG125. We made new primers based on the Cre sequence given to us from Griffitts labs.
This is the sequence for Cre from the Griffitts lab in pJG125
Once we PCR amplify this gene, we can clone it into the Lox plasmid that we have created.
5/8/2012 EJ and HE
Analysis of PCR products by agarose gel electrophoresis:
We ran a gel on agarose gel to assess the quality of our Cre product.
- Joe made a standard 1% gel (not low-melt) of 1XTAE and 0.75g of agarose. Put it into the microwave for about 90s until the agarose completely dissolved.
- Added 12μL of 1mg/mL ethidium bromide and swirled to mix. Wore gloves.
- Allowed the flask to cool so that the glass felt warm, not burning hot. Poured the liquid onto the gel bed and let it cool. Inserted the appropriate sample comb.
- Added loading dye to Cre PCR product. Added 6μL of 10X loading dye to the PCR tube.
- Moved the gel into the proper orientation in the gel box, covered the gel with 1X TAE buffer and loaded 5μL Cre PCR product into well 11.
- Turned on power suply to 100V. Ran for 40-60min. Results are below:
Our Cre didn't work because we froze the phusion polymerase. We had to start with the PCR of Cre again from 5/7/2012.
5/11/2012 HE and EJ
Reran the Cre PCR and then run it out on a gel.
The cre gel didn't work, the last two wells were cre control and cre-3 which are labelled CC and C3. It looks like the cre-3 (3 because it's try #3) got stuck in the well somehow, so we are going to run the gel again at the beginning of the day on Monday, and decide what to do from there if it doesn't work.
The picture is on the E.Colinoscopy USB titled "audrey and Brooke" and the gel details are in Audrey's notebook.
5/15/2012 HE and EJ
Ran the gel with CC and C3 again and:
It did NOT work! :( :( :( .
So we are going to start over again with the Cre (Try #4). I boiled template today by adding 50μL water to a PCR tube and picking up a colony of pJG125 with a pipette tip and depositing it in the PCR tube. Then I boiled the colony in the PCR machine to get out the DNA. Now we will run PCR on the Cre gene again.
PCR amplify Cre gene
35 ul ddH2OH2O
10 ul Hf (5x) buffer
1.5 ul 10mM dNTP
1 ul plasmid (pJG125 which was boiled last time by Hilary)
1 ul B155 (forward primer)
1 ul B156 (reverse primer)
.5 ul Phusion polymerase
50 ul total
This time we added the water first, which we did not do last time. Maybe that was the problem. The last few times we added the water just before the phusion polymerase, so that may be why it wasn't working.
We labelled the tube with template "C4" and the control "CC". We put them in the PCR machine overnight on Phusion PCR cycle. Hopefully it works this time!!!!
We labelled the template "CT4" and put it in the colon cancer box in the freezer
Future
Now that we know the PCR worked, we did a PCR product purification to clean up our Cre gene.
The Cre gene is now ready for restriction digest. We will also perform restriction digest on our Lox plasmid so that we can clone the Cre gene into the Lox plasmid.
5/16/2012 HE
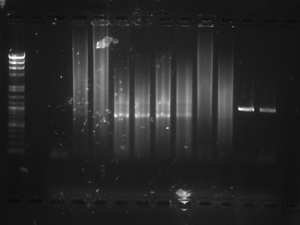
We ran our cre on a normal gel and the last two wells in the picture above are the results of cre. As you can see above now our plans are to purify our cre plasmid and then do a restriction digest on both cre and lox to put them together.
5/18/2012 HE
We purified the cre PCR and it is in the colon cancer box labelled "PURIFIED DNA CRE 5/18/2012"
(the directions to purify the PCR are missing from th QIAGEN PCR purification kit and so they are located here: http://www.qiagen.com/literature/default.aspx?Term=PCR+purification&Language=EN&LiteratureType=1&ProductCategory=230 under "QIAquick spin handbook"
On Monday we need to work on doing the restriction digest and low melt gels for both cre and lox to prepare to clone both of them together. Making progress! Feels good!
5/21/2012 HE
Figured out what enzymes are necessary for the Restriction digest and low-melt gel (HindIII HF and PstI)
5/22/2012 HE
Did restriction digest on the purified cre DNA as well as the purified lox DNA. labelled the tubes Vector lox RD and PCR Prd. Cre RD. Used the previously mentioned enzymes (HindIII and PstI) After incubating them for 1.5 hours at 37C and letting making the low-melt gel, I inserted in this order to the wells
14μL ladder 55μL Purified Cre PCR product 55μL purified lox plasmid (vector)
after running the low-melt gel for 60 minutes I looked at it under the UV light and there was NO Cre PCR band ONLY lox. So I have no idea what happened to the DNA. Dr. Grose said it must have happened during the purification process.
We have another tube of Cre PCR that was ran on the gel and worked, so we need to purify that PCR product, do a restriction digest, and run it on a low-melt gel again. I put the low-melt gel product from lox in the freezer (yellow container) and it is labelled "lox 5/22"
5/29/2012 EJ and KN
Because Cre did not work, we needed to do the PCR again. Hopefully this will be the final time.
First I boiled template, which came from the pJG125 plasmid in the refrigerator.
PCR amplify Cre gene
35 ul ddH2OH2O
10 ul Hf (5x) buffer
1.5 ul 10mM dNTP
1 ul plasmid (pJG125 which was boiled last time by Hilary)
1 ul B155 (forward primer)
1 ul B156 (reverse primer)
.5 ul Phusion polymerase
50 ul total
I labeled the cotrol CC5 and I labeled the real PCRs C5 with a 1 on the side, C5 with a 2 on the side, and C6. I had to make 3 different tubes because the first 2 tubes, I forgot to mix the PCR mix before adding the phusion polymerase. So hopefully all 3 PCR tubes work, but C6 should work for sure.
Then I ran the PCRs on a phusion polymerase cycle. After it was finished, I put the PCR tubes in the E. colinoscopy box in the freezer.
5/30/2012 HE
Ran the PCR product out on a regular gel: It was as follows from well 1-well 5 LADDER CC5 C5-1 C5-2 C6-3 There was a band in all 4 of the PCR product wells, including the control. I talked to Jordan about why this would be, because we had a band in our control last time as well, and he said it could be contaminated reagents. (some DNA was added to the reagents and that is what we see) He said that that is a possibility but highly unlikely. We are going to go forward with these results and if we don't have any product in our low-melt again then we will go ahead and run the PCR again with all new/different reagents to illuminate contamination potentials.

June
6/4/2012 EJ and HE
PCR Cleanup
In this step, we used C5-2 and C6. Eric did C5-2 and Hilary did C6. Hopefully one of these will work.
- Added 250μL of Buffer PB the PCR product.
- Placed a QIAquick spin column in a provided 2 ml collection tube.
- To bind DNA, applied the sample to the QIAquick column and centrifuged for 30–60 s.
- Discarded flow-through. Placed the QIAquick column back into the same tube.
- To wash, added 0.75 ml Buffer PE to the QIAquick column and centrifuged for 30–60 s.
- Discarded flow-through and placed the QIAquick column back in the same tube. Centrifuged the column for an additional 1 min.
IMPORTANT: Residual ethanol from Buffer PE will not be completely removed unless the flow-through is discarded before this additional centrifugation.
- Placed QIAquick column in a clean 1.5 ml microcentrifuge tube.
- To elute DNA, added 50 μl Buffer EB (10 mM Tris·Cl, pH 8.5) to the center of the QIAquick membrane. Let stand for 3 min and centrifuged the column for 1 min.
IMPORTANT: Ensure that the elution buffer is dispensed directly onto the QIAquick membrane for complete elution of bound DNA. The average eluate volume is 48 μl from 50 μl elution buffer volume, and 28 μl from 30 μl elution buffer. Elution efficiency is dependent on pH. The maximum elution efficiency is achieved between pH 7.0 and 8.5.
Restriction Digest of Insert
We did this procedure with C5-2 and C6.
14μL ddH2O
5μL 10X NEB buffer
0.5μL 100X BSA
30μL DNA sample
1-2μL of each restriction enzyme
After adding these to PCR tubes labeled C5-2 and C6, we put the tubes in the incubator for 1.5 hr.
Low Melt Gel - we ran them on the low-melt gel and they showed up, however it is important to note that the C5-2 seemed to be a little smaller/didn't run as far down as the C6. We have no explanation for this except weird. I am putting them in the colon cancer box. They are labelled C5 and C6 with today's date (6/4/2012) on the side.
6/5/2012 HE and EJ
Ligation
With insert--PCR tubes "CL5" and "CL6" (underlined and superlined)
- 6.5 μL ddH2O
- 1.5 μL 10X ligase buffer (includes ATP)
- 1μL T4 DNA ligase
- 3μL vector
- 3μL insert
Control (w/o insert) -- PCR tube labeled "CLC"
- 6.5 μL ddH2O
- 1.5 μL 10X ligase buffer (includes ATP)
- 1μL T4 DNA ligase
- 3μL vector
- 3μL insert
The water, ligase buffer and ligase were combined as a master mix and transfered into PCR tubes for the ligation. Then the vector was added to both tubes and the insert was added to only one of the tubes. The control tube was labeled "CLC" and the tube with insert was labeled "CL". The tubes were incubated at room temperature for 80 minutes from 11:30-12:50.
The low melt gel tubes were labeled with "LM" for low-melt.
Transformation
Protocol:
- Thawed DH5α cells on ice. Melted the ligations at 65°C.
- Added 2μL of ligation mix to about 25μL of competant cells. Flicked the tubes briefly and put them back on ice for 5-10 min. Labelled the tubs "ET C52" "ET C63" and "ET CC".
- Heat shocked at 42°C for 60 sec. Immediately put the tubes back on ice for 2-5 min.
- Added 500μL of plain LB to the rxns and incubated at 37°C for 35 min from 1:15-1:50.
- Plated all 500μL of cells and incubated at 37°C overnight.
The 500μL plates that will be in the 37C overnight are labelled crelox transformation control, crelox transformation C5-2, and crelox transformation C6-3 they all have the date on them 6/5/2012 and need to be taken out tomorrow.
6/5/2012 EJ
Colony PCR
Put colonies in 8-tube PCR tubes along with 50μL ddH2O. I labelled the colonies 1-8 on the plates and made sure to put them into the corresponding labelled PCR tube. Boiled the template and froze it. It is ready for PCR next time.
6/11/2012 EJ
Set up the Taq PCR reaction:
Made a master mix (enough for 16 rxns)
- 304μL ddH2O
- 40μL Standard 10X rxn buffer
- 8μL 10 mM dNTP's
- 8μL IG57 primer
- 8μL BI12 pPLAT reverse primer
Mixed well before adding
- 8μL Taq polymerase
Then I transferred 23μL of the master mix to each PCR tube. Then added
- 2μL of boiled colony sample
Put the two 8 PCR tube sets into the PCR machine on a Taq PCR cycle. It should be finished by 4:00pm today. Then I will need to run a gel to ensure that the Cre gene made it into the insert.
6/12/2012 HE

As you can see we do have product from our colony PCR but after looking at the sizes it is clear that these PCR's only contain lox and cre has still not been cloned in. We will try again! Starting back with the Cre PCR and not making a control because our primers are clearly contaminated since our control consistently has product in it.
Argggggggggggggggggggggggggggggggggggg!
Next step: PCR Cre
6/18/2012
PCR amplify Cre gene
35 ul ddH2OH2O
10 ul Hf (5x) buffer
1.5 ul 10mM dNTP
1 ul plasmid (pJG125 which was boiled last time by Hilary)
1 ul B155 (forward primer)
1 ul B156 (reverse primer)
.5 ul Phusion polymerase
50 ul total
I labelled the tube Cre PCR. Put it in the Cre PCR machine at 1:20.

Overnight Poured Ara/Amp broth into a test tube and then added the Lox plasmid transformed bacteria from slice 1 of plate 1. Set it in the 37°C incubator overnight.
6/19/2012
PCR Cleanup
- Added 250μL of Buffer PB the PCR product.
- Placed a QIAquick spin column in a provided 2 ml collection tube.
- To bind DNA, applied the sample to the QIAquick column and centrifuged for 30–60 s.
- Discarded flow-through. Placed the QIAquick column back into the same tube.
- To wash, added 0.75 ml Buffer PE to the QIAquick column and centrifuged for 30–60 s.
- Discarded flow-through and placed the QIAquick column back in the same tube. Centrifuged the column for an additional 1 min.
IMPORTANT: Residual ethanol from Buffer PE will not be completely removed unless the flow-through is discarded before this additional centrifugation.
- Placed QIAquick column in a clean 1.5 ml microcentrifuge tube.
- To elute DNA, added 50 μl Buffer EB (10 mM Tris·Cl, pH 8.5) to the center of the QIAquick membrane. Let stand for 3 min and centrifuged the column for 1 min.
IMPORTANT: Ensure that the elution buffer is dispensed directly onto the QIAquick membrane for complete elution of bound DNA. The average eluate volume is 48 μl from 50 μl elution buffer volume, and 28 μl from 30 μl elution buffer. Elution efficiency is dependent on pH. The maximum elution efficiency is achieved between pH 7.0 and 8.5.
Restriction Digest of Insert and Plasmid
14μL ddH2O
5μL 10X NEB buffer
0.5μL 100X BSA
30μL DNA sample
1-2μL HindIII
1-2μL PstI
After adding these to PCR tubes labeled "Cre Ins" and "Lox", we put the tubes in the 37°C incubator for 1.5 hr starting at 12:10.
I should note that we are having doubts as to whether our Lox plasmid was done correctly. In our protocol we wrote that for the restriction digest of Lox we used EcoRI, but that would have been incorrect. We should have used HindIII. We are going to resequence our Lox plasmid and hopefully get better results. If there was a mix-up with restriction enzymes that could be why we have been having so much problems getting Cre into the plasmid. If EcoRI had been used, the HindIII site would have been removed.
We sent off our lox plasmid to the sequencing center again. We want to make sure that lox is actually in the plasmid and that it is between the correct restriction sites. Added 1.5μL plasmid to two PCR tubes and then 1μL of BI12 to one tube and 1μL of IG57 to the other tube.
Low Melt Gel
Mixed 0.8g low melting temperature agarose with 75ml TAE buffer in a flask. Microwaved for three 20 second periods and swished to mix in between periods. Waited for the mixture to cool down so that it wasn't scalding hot. Added 1 drop ethidium bromide and mix. Poured gel and used the large comb.
Cut out the band for our insert and the band for our plasmid. Made sure to not get too much extra gel in order to keep the DNA concentration high in the gel cut out.
Results: Cre came out to be about 1-1.5 kb (very large) and the part of the lox plasmid which was cut out was about 600base pairs which is a pretty large chunk, so I'm not sure why that would be. We need to discuss these results with Dr. Grose and press forward with the ligation tomorrow. The tubes are marked well 2 lox and well 4 cre and are in the cre-lox box in the freezer.
we also didn't get any results for the overnight and so we need to take those out tomorrow.
6/20/12
Take out the overnight!
6/21/12
well... we tried to start the ligation, but soon realized that it wasn't a low-melt gel that was used and so we are not able to melt the gel to ligate them. So we started the cre PCR again and also purified the Lox plasmids so we can have more to work with when we get back to the low-melt step again.
We need to purify the cre PCR as soon as it's done! The Cre template is in the cre-lox box as well as the two gel slices which are not low melt, we need to ask Dr. Grose if there is a way to extract the DNA from the non-lowmelt gels.
6/27/2012 EJ
Dr. Grose got back the sequencing results for Lox again. We looked to see where it cloned in, and it was not between the cut sites we expected it to be. How in the world it got somewhere else we have no idea. It's really strange. Take home message is that we need to re-clone lox into our plasmid. Since we already have Cre PCR'd and ready to go we may as well clone that into a new plasmid, and then get lox into the plasmid. Let's do it!
Restriction Digest of Plasmid
14μL ddH2O
5μL 10X NEB buffer
0.5μL 100X BSA
30μL pIG12
1.6μL HindIII HF
1.6μL PstI HF
After I added everything together, I put them in the 37°C water bath from 3:45 to 8:20. I labelled the tube PIG12 RD and I put it in the freezer.
6/28/2012 EJ and HE
Hillary re-PCR'd Cre and purified it.
Poured a low-melt gel in to run out the PIG12 RD. Ran the low-melt from 12:30-1:20.
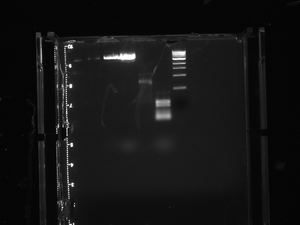
Put the gel slice in the freezer in the Cre Lox box. It is labelled "PIG12 Low-melt". I put both of Audrey's gel slices in the Cholera box in the freezer.
Started Lox. Boiled bacteria from pJG181, and streaked a plate for future use. I put the plate in the 37°C incubator, so I will need to take it out tomorrow.
Checked the gel with the Cre PCR on it around 12:05. Print two pictures, one for Justin. Key: Ladder, 2 lanes with Justin's stuff, Cre.
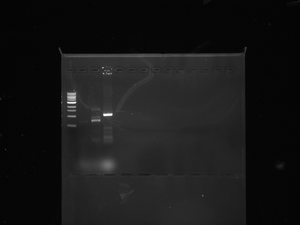
Dr. Grose looked at this gel and said that Cre looked fine. So we will continue to the RD with Cre
pCONLOXTERMLOX PCR
This is the PCR setup to amplify pCONLoxTermLox from pIG181. These primers should be correct.
- 35μL ddH2O
- 10μL 5X Phusion buffer
- 1.5μL 10mM dNTP's
- 1μL Primer BI133 (SacI cut site)
- 1μL Primer IG84 (HindIII cut site)
- 0.5μL Phusion Polymerase
I labelled the tube "Lox PCR". It is in the PCR machine.
Cre Restriction Digest
I added the following to the purified Cre. I omitted the water and DNA sample, because that is what Dr. Grose said to do. I had a little trouble getting the BSA buffer out of the pipet tip because it was such a small amount, so if there is a problem, that might be the source.
- 5μL 10X NEB buffer
- 0.5μL 100X BSA
- 1.6μL HindIII HF
- 1.6μL PstI HF
Incubated mixture into the 37°C bath for 2.5hrs from 2:00-4:30. I then put it in the Cre Lox box in the freezer. It is labelled "Purified Cre DNA".
Poured a low melt gel to run out the Cre on as soon as the RD is finished. Put it in the refrigerator for use tomorrow.
July
7/2/2012
We ran the lox PCR out on a gel and then did the PCR cleanup, unfortunately neither of them worked out! So we are running the PCR again:
pCONLOXTERMLOX PCR
This is the PCR setup to amplify pCONLoxTermLox from pIG181. These primers should be correct.
- 35μL ddH2O
- 10μL 5X Phusion buffer
- 1.5μL 10mM dNTP's
- 1μL Primer BI133 (SacI cut site)
- 1μL Primer IG84 (HindIII cut site)
- 0.5μL Phusion Polymerase
I labelled the tube "Lox PCR". It is in the PCR machine.
Ran the low melt gel. The Cre looks to be about the right size (just over 1kB). Here is a picture:
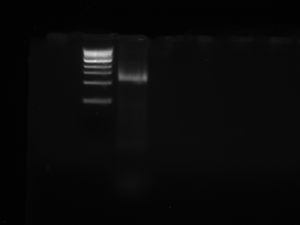
Cre Ligation into pIG12
Melt the gel slices at 65°C
- 6.5μL ddH2O
- 1.5μL 10X ligase buffer
- 1μL T4 DNA ligase
- 3μL vector (pIG12)
- 3μL insert (Cre)
Incubated at room temperature for 45 min from 4:40-5:25.
Transformation
- Thawed DH5α cells on ice. Melted the ligations at 65°C.
- Added 2μL of ligation mix to about 25μL of competant cells. Flicked the tubes briefly and put them back on ice for 5-10 min. Labelled the tube "ET Cre"
- Heat shocked at 42°C for 60 sec. Immediately put the tube back on ice for 2-5 min.
- Added 500μL of plain LB to the rxn and incubated at 37°C for 45 min.
- Plated all 500μL of cells and incubated at 37°C overnight.
The 500μL plates that will be in the 37°C overnight are labelled Cre transformation CONTROL and Cre transformation. Both have the date on them (7/2/2012) and need to be taken out tomorrow.
7/3/2012 EJ
Colony PCR
I chose 8 colonies from the Cre Transformation plate to make template for TAQ colony PCR, and streaked them out on a plate for future use. Put the plate in the 30°C incubator for two days, until Thursday. Set up the TAQ PCR rxn with Jordan. Made a master mix:
- 190μL ddH2O
- 25μL standard 10x rxn buffer
- 5μL 10mM dNTP's
- 5μL BI12 pLAT reverse primer
- 5μL IG57 primer
Mixed these reagents well and then added
- 5μL TAQ DNA polymerase
Then we added 2μL of boiled template to each well, making sure to keep the numbering the same (the template from one went into the tube labelled 1, 2 into 2, etc). Ran the TAQ PCR overnight.
7/5/2012
Ran my TAQ PCR out on a gel. My lanes were the ones in the middle of the gel. I loaded ladder, wells 1-8, and then the control. Here is a picture:
![]()
As you can see, there is absolutely no DNA in our lanes. Wo is me!!!
We figured that a plasmid had to make it into the bacteria because colonies grew on the plate which had ampicillin in it. So there may have been a problem with the template that I used. We re-ran the TAQ PCR. We made the following master mix:
- 190μL ddH2O
- 25μL standard 10x rxn buffer
- 5μL 10mM dNTP's
- 5μL BI12 pLAT reverse primer
- 5μL IG57 primer
Mixed these reagents well and then added
- 5μL TAQ DNA polymerase
Then we added 2μL of boiled template to each well, making sure to keep the numbering the same (the template from one went into the tube labelled 1, 2 into 2, etc). Ran the TAQ PCR overnight.
Update: This colony PCR showed that the insert did not make it into any of the plasmids. Start over.
7/10/2012
Boiled template for Cre. This was done with bacteria pJG125.
Dr. Grose ran the following PCR with Cre.
- 35μL ddH2O
- 10μL 5X Phusion buffer
- 1.5μL 10mM dNTP's
- 1μL Primer BI133 (SacI cut site)
- 1μL Primer IG84 (HindIII cut site)
- 0.5μL Phusion Polymerase
Ran the PCR out on a gel. These were the results. Cre is in the top lanes. From right to left it goes: ladder, Cre, Cre control.
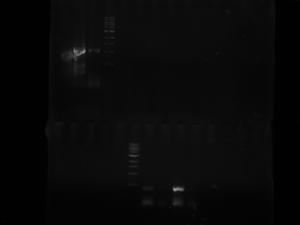
Eric ran the PCR cleanup along with Justin and Joseph.
Dr. Grose ran the following Restriction Digest:
- 50μL DNA (pIG12 or Cre cleanup)
- 5μL 10X NEB buffer
- 0.5μL 100X BSA
- 1-2μL PSTI HF
- 1-2μL HINDIII HF
Left in the water bath at 37°C from 1:00-3:30. Poured a low melt gel. Then Hillary ran the low melt with Cre, PJG125, and Joseph/Justin's PCR stuff. Cut out the gel and put into tubes labelled "pJG125 LM" and "Cre LM".
7/16/2012
It appears that the Cre transformation worked!
We ran the following TAQ PCR on the Cre colonies. We made the following master mix:
- 190μL ddH2O
- 25μL standard 10x rxn buffer
- 5μL 10mM dNTP's
- 5μL BI12 pLAT reverse primer
- 5μL IG57 primer
Mixed these reagents well and then added
- 5μL TAQ DNA polymerase
Then we added 2μL of boiled template to each well, making sure to keep the numbering the same (the template from one went into the tube labelled 1, 2 into 2, etc). Ran the TAQ PCR overnight.
This is the PCR setup to amplify pCONLoxTermLox from pIG181. These primers should be correct, according to Dr. Grose. If they are correct, I don't know what we were doing wrong before, because these were the same primers we used. This is the IG84 sequence from the primer tube: CCG AAG CTT TTT ACG GCT AGC TCA GTC CTA GGT ATA GTG CTA GCC ATA ACT TCG TAT AGC.
- 35μL ddH2O
- 10μL 5X Phusion buffer
- 1.5μL 10mM dNTP's
- 1μL Primer BI133 (SacI cut site)
- 1μL Primer IG84 (HindIII cut site)
- 0.5μL Phusion Polymerase
I labelled the tube "Lox PCR". It is in the PCR machine.
7/26/2012
This is the PCR setup to amplify pCONLoxTermLox from pIG181. These primers should be correct.
- 35μL ddH2O
- 10μL 5X Phusion buffer
- 1.5μL 10mM dNTP's
- 1μL Primer BI133 (SacI cut site)
- 1μL Primer IG84 (HindIII cut site)
- 0.5μL Phusion Polymerase
I labelled the tube "L" and the control "LC". They are in the PCR machine. Next step is to do a gel.
