Chris Rhodes Week 3
Methods and Results
Activity 1 Parts 2 and 3
- Following the Genbank links under the Secondary Source ID tab from the PubMed article I added the Fasta sequences of AF016760, AFO016761, AFO016767, AFO016780, and AFO016787 to the WorkBench by clicking the Add Sequence button under Nucletotide Tools section and copying and pasting the FASTA sequence into the upload box.Then by selecting all the uploaded sequences and clicking the ClustalW button under Nucleotide Tools I ran a ClustalW multiple sequence allignment using the default parameters: (Note: All ClustalW and Clusdist alignments were performed using the default parameters)

The blue regions of the alignment represent where the nucleotides of the different sequences all match each other while the black regions represent an area where one nucleotide of at least one sequence differs from the nucleotides of the other sequences in the alignment.
- I also looked at the unrooted genetic tree and took a screenshot:

The genetic tree shows the relative difference between all the sequences in the alignment through the use of distance on the tree. The closer the sequences are to each other the more similar the genetic sequences are to each other.
Activity 2 Part 1
- I added all of the nucleic acid sequences of both .txt files onto the Workbench by uploading each .txt file into the Nucleotide Tools section of Workbench. The .txt files can be found here HIV-1 In-Class Activity Page under the background section along with a link to the Activity handout and the general requirements of the assignment.
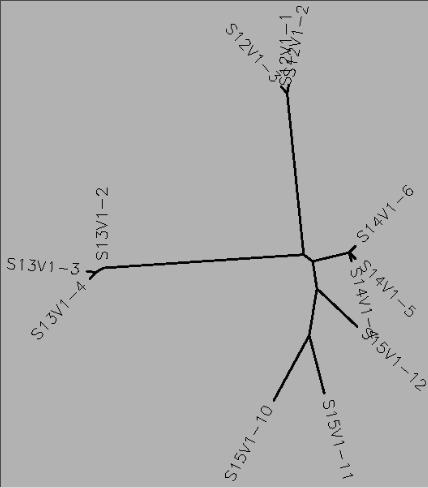
- From the sequences I ran a ClustalW allignment of: S12V1-1, S12V1-2, S12V1-3, S13V1-2, S13V1-3, S13V1-4, S14V1-4, S14V1-5, S14V1-6, S15V1-10, S15V1-11, S15V1-12 and took a screenshot of the alignment and the unrooted genetic tree:



Activity 2 Part 2
- To calculate the S value of a particular alignment I selected all of Subject 8's clones and ran a ClustalW alignment. The S value corresponds to the number of nucleotide sites that differ between any of the sequences aligned. For our purposes the S value can be found by counting the number of black columns throughout the ClustalW alignment. For example, the S value of the alignment below is 6.

- An alignment was also done for Subject 9

- and Subject 10

- From these alignments and using the equations given in the Activity Handout S and Theta were calculated.
- By using the equations given in the Activity Handout and the Clustdist tool under the Alignment Tools section for each alignment the Min and Max differences were determined. (See Single Subject Alignment Table below)
- Clustdist of Subject 8

- Clustdist of Subject 9

- Clustdist of Subject 10

Activity 2, Part 2: Single Subject Alignment Table
Subject Clones S Theta Min-Diff. Max-Diff. 8 5 6 2.88 1.14 5.13 9 5 5 2.44 1.99 3.99 10 7 7 2.86 1.14 3.14
- Using Subjects 12 and 13 I created and saved a new ClustalW alignment to check the Min and Max differences across subjects by using Clustdist.
- Subject 12 and 13 Alignment

- Subject 12 and 13 Clusdist

- This was repeated using a Subject 8 and 9 alignment and a Subject 8 and 10 alignment.
- Subject 8 and 9 Clustdist

- Subject 8 and 10 Clustdist

- Using the Clustdist of all the cross subject alignments the Min and Max differences for each were calculated. (Below)
Activity 2, Part 2: Cross Subject Alignment Table
Subject Crosses Min-Diff. Max-Diff. 12 and 13 39.05 43.89 8 and 9 27.93 33.06 8 and 10 27.1 31.07
Activity Questions
Activity 1, Part 2: GenBank
- AF016760, AFO016761, AFO016767, AFO016780, and AFO016787
- These gene sequences all came from subject 1
Activity 2, Part 1
- Clones from each subject tend to cluster together on the unrooted genetic tree
- Some subject clusters are much closer to each other on the tree than others specifically Subjects 13,14,and 15
- From the pattern of clustering seen in the genetic tree it seems that the genetic tree separates individual sequences based on their similarity to each other. The closer one sequence is to another on the tree, the more closely those two sequences are related to each other in terms of sequence similarity or evolutionary time.
Markham Paper Prep
- Vocab List
- Seroconvertion: The process by which the body begins to produce specific anitbodies to fight a specific infection or disease in the blood.
- Sanger chain termination: A method of DNA sequencing in which ddNTP's are used in PCR to terminate the transcription of DNA at various sites resulting in DNA strands of varying length which can be run using high sensitivity gel electrophoresis and interpreted to determine the sequence of a DNA sample.
- Progressor: A term used to describe the varying degrees of HIV-1 progression in patients through the measure of decline of CD4 T cells.
- Chemostat: A bioreactor in which fresh medium is constantly add while cultured medium is constantly removed in order to maintain a constant volume of culture in the reactor.
- Cohorts: A group of organisms of the same species that all share similar characteristics which are studied over time.
- Peripheral blood mononuclear cells: Any blood cell that contains a round nucleus. CD4 T cells are peripheral blood mononuclear cells.
- Plasma: Fluid in which the various components of blood, including proteins, glucose, clotting factors, and hormones, are suspended
- External Primers: Secondary primers used in PCR in order to isolate unwanted gene sequences and serve as buffers between the unwanted sequences and the nested primers.
- Nested Primers: Primary primers used in PCR in order to more specifically select amplification of a desired region without the risk of outside contamination of any unwanted gene sequence. More detailed information about Nested PCR can be found here Nested PCR Information
- Synonymous mutations: Mutations in which the change of the nucleotide does not effect the amino acid produced after translation.
Outline
Introduction
- HIV-1 viruses have a high mutation rate, which contributes greatly to its ability to rapidly adapt to selective pressure in an environment.
- It is important to observe the genetic diversity and specific mutations of HIV-1 viruses during their evolution in order to better understand both the methods and efficiency of the selective forces working against the viruses and the manner in which the viruses overcome those forces.
- Previous studies involved too small of a subject group, used techniques that did not utilize the specific genetic sequences of each virus, and failed to use enough time points to be considered accurate and thorough accounts of the viral progression.
- The paper shows two things, 1) Different selective pressures are associated with the different rates of CD4 cell decline 2) The greater number of genetic variants in a system, the more rapid the CD4 cell decline.
Methods
- The study employed the use of 15 subjects who were placed in groups (Rapid, Moderate, and Nonprogressors) based on their rate of CD4 cell decline.
- The genetic sequence of each virus variant from each subject for each visit was determined through the used of nested PCR and each sequence was uploaded onto GenBank for future use.
- Phylogenetic Trees for the all the variants of the various subjects were created using the Mega computer package
- The rates of synonymous and nonsynonymous mutations were calculated from the genetic sequences and the data was corrected for bias by adjusting for the number of possible mutation sites for each mutation type.
- Diversity was calculated as the mean of the number of nucleotide differences between clones
- Divergence was calculated as the mean of the percent difference of the nucleotides of the clones from a consensus sequence taken from one of the first visit clones.
- Rates of diversity and divergence were calculated using regression lines tracing all the diversity and divergence data points for each subject across the entire experimental timeline.
Explanation of Figures/Results
- Figure 1 shows the progression of T cell count, diversity, and divergence for all 15 subjects as a function of time since their first testing visit.
- Rapid progressors show the fastest rate of T cell count decline, along with an overall increase in both diversity and divergence expect for subject 15 who experienced a net drop in diversity.
- Moderate progressors all show a net decline in T cell count except for subject 6 who shows a small increase however, diversity and divergence increased for all moderate progressors.
- Nonprogressors all show net increases in T cell count, diversity, and divergence though the change in diversity and divergence is minimal.
- Table 1 shows all the data points collected and calculated throughout the experiment. These include number of visits, original CD4 counts, diversity, viral clone count, rate of CD4 decline, rate of change of diversity, rate of change of divergence, and dS/dN.
- Rate of CD4 Decline: The table shows that the rate of CD4 decline decreases from Rapid to Moderate to Nonprogressors.
- Rate of Change of Diversity: The table shows how quickly variant change occurred for each subject and that for all subjects except for 7 and 15 there was a net increase in viral diversity over time.
- Rate of Change of Divergence: The table shows how quickly the various subject’s clones changed from the original consensus sequence and that for all subjects there was a net increase in divergence over time.
- dS/dN: A dS/dN value less than one means that there is selective pressure favoring change in the amino acid sequence, a dS/dN value of 1 means there is no selective pressure for or against change in the amino acid sequence, and a dS/dN value of greater than 1 means that there is selective pressure against change in the amino acid sequence.
- Figure 2 shows the mean rate of change of diversity and divergence for the 3 different types of progressors.
- The rate of change of diversity seems to increases from nonprogressor to moderate progressor to rapid progressor however there are large error bars for the rapid progressor group that may intersect with the error bars for the moderate progressors. Difference between the two groups was shown to not achieve significance (P=.17)
- The rate of change of divergence would seem to increase from nonprogressor to moderate progressor to rapid progressor however the error bars of the rapid progressor group intersect with the error bars of the moderate group calling this relationship into question. Difference between the two groups was shown to not achieve significance (P=.08)
- Figure 3 shows the phylogenetic tree of all of subject 9’s cataloged clones with the distance between the clones of each visit showing increasingly larger divergence over time from the original clones.
- Figure 4 shows multiple phylogenetic trees including subjects 5, 7, 8, and 14. Each tree shows the genetic evolution of each subject’s virus clone and subsequent clones vary greater and greater over time.
Discussion
- Higher diversity and divergence corresponds to a greater rate of CD4 decline.
- Synonymous mutations were not favored in any progressor group, but nonsynonymous mutations were more abundant in rapid and moderate progressors than in nonprgoressors. This indicates that there is selection for change in the envelope sequence of rapid and moderate progressors and selection against change in the nonprogressors.
- This conclusion contradicts the established “best fit” model of virus proliferation.
- This report agrees with the Mcdonald study that viral strains in rapid progressors show greater genetic divergence, but disagrees with McDonald’s finding that rapid progressors had less net genetic diversity than slow progressors.
- It is hypothesized that the difference in the two studies is the result of insufficient time points in Mcdonald’s study.
- As with Mcdonald’s report, this paper disagrees with Wolinsky’s study showing decreases genetic diversity in rapid vs slow progressors.
- It is hypothesized that this difference can be attributed to an exceptional subject whose immune response was not adequate enough to mount a proper offensive.
- This report agrees with the Nowak study stating that increasing genetic diversity is associated to increasing CD4 cell decline rates.
- It can be hypothesized that a main difference between rapid progressors and nonprogessors is the specificity of their immune systems. Rapid progressors would seem to have specifically targeted immune responses to specific viral variants where as nonprogressors would seem to attack the variants more broadly thus more effectively cutting down genetic diversity.
Links
- Chris Rhodes User Page
- Week 2 Journal
- Week 3 Journal
- Week 4 Journal
- Week 5 Journal
- Week 6 Journal
- Week 7 Journal
- Week 8 Journal
- Week 9 Journal
- Week 10 Journal
- Week 11 Journal
- Week 12 Journal
- Week 13 Journal
- Week 14 Journal
- Home Page
- Week 5 Assignment Page
- Week 6 Assignment Page
- Week 7 Assignment Page
- Week 8 Assignment Page
- Week 9 Assignment Page
- Week 10 Assignment Page
- Week 11 Assignment Page
- Week 12 Assignment Page
- Week 13 Assignment Page
- Week 14 Assignment Page
