BISC 219: Mod 2 Lab 5

Lab 5: The pL4440 Plasmid
Plasmids are circular pieces of DNA that can replicate in bacteria but are not part of the bacterial chromosome. Plasmids are generally circular molecules with fewer base pairs of DNA than the chromosome and with certain sequence elements (called the origin or ori) that allow the plasmid to replicate within the bacterial cytoplasm. Many naturally occurring plasmids have been modified for the purposes of using them as research tools. For example, a gene encoding resistance to an antibiotic can be added to a plasmid so that bacteria carrying the plasmid will become antibiotic resistant. This modification allows for selection of cells that carry plasmid DNA. A simplified map of the C. elegans RNAi plasmid is below:
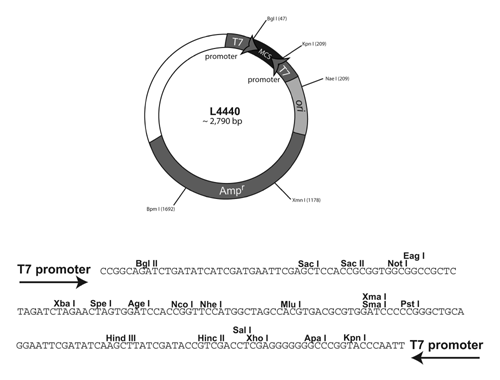
To enable us to make lots and lots of RNA for RNA interference we need to express our gene at high levels. This is done with a specific strain of E. coli called HT115(DE3). The bacteria cells contain the T7 RNA polymerase gene (contained within a stable insertion of a modified lambda prophage λ DE3) under the control of lac operon regulatory elements. This allows expression of T7 polymerase to be controlled by isopropyl-β-D-thiogalactopyranoside (IPTG), a lactose analogue that induces expression of genes under the control of the lac operon o gene. When IPTG is added, the cells will begin to synthesize lots of T7 RNA polymerase. This T7 RNA polymerase can then bind to the T7 promoter sites on the plasmid and begin to synthesize RNA from both T7 RNA polymerase sites. Because the two single strands of RNA are complementary to each other they will form double stranded RNA within the bacterial cell. Additionally, this particular strain is deficient for the RNAaseIII enzyme that degrades double stranded RNA (dsRNA) in the bacterial cell. This allows for the accumulation of dsRNA in the cell and, thus, our ability to induce and RNAi effect! This E. coli strain carries a tetracyclin resistance gene so these cells can be selected on media containing tetracyclin, while the plasmid contains an ampicillin resistance gene that allows only transfomed cells to grow on media containing ampicillin.
Background Reading on Bacterial Transformation
During “transformation,” a single plasmid enters a single bacterium and, once inside, replicates and expresses the genes it encodes. In this case, the relevant genes expressed are for ampicillin resistance and for the piece of the C. elegans gene of interest. The transformation mixes were given a short time to express these gene products and then were spread on an agar plate that contained nutrients and the antibiotics tetracyclin (encoded by the bacteria) and ampicillin (encoded by the plasmid). Only the cells that incorporated the plasmid DNA and expressed the plasmid genes grew to form colonies of bacteria in the presence of ampicillin. The untransformed bacteria failed to form visible colonies on the ampicillin containing agar surface.
Most bacteria do not usually exist in a “transformation ready” state, but the bacteria can be made permeable to the plasmid DNA by exposing them to calcium chloride. Cells that have been treated with calcium chloride or are otherwise capable of transformation are referred to as “competent.” Competent cells are extremely fragile and must be handled gently, i.e. kept cold, not vortexed, etc. The transformation procedure is efficient enough for most lab purposes; with efficiencies as high as 107 transformed cells per microgram of DNA, but it is important to realize that only 1 cell in about 10,000 is successfully transformed.
Plasmid Isolation and Transformation
Last week, you selected a colony of bacteria that contains a plasmid with a specific C. elegans gene you will RNAi. Now you will isolate the plasmid DNA and transform it into a HT115(DE3)strain for two reasons: we can induce the HT115(DE) strain to make lots of our gene product of interest and because a freshly transformed culture seems to work much better for eventual knockdown of gene expression by dsRNA interference. You may be wondering why we are taking the plasmid out of E. coli and then putting it back into another fresh culture of E. coli. There is a lot scientists don't yet know about the mechanism of dsRNA interference. One thing we do know is that RNAi knockdown works better when you use freshly transformed feeding bacteria that can be manipulated to overexpress plasmid genes of interest, so that's what we'll do.
Yesterday you inoculated a few milliliters of LB broth containing bacteria maintaining a genetically engineered plasmid, pL4440, that has been modified to contain an antibiotic resistance gene to ampicillin and all or part of a C. elegans gene that you want to investigate. You added ampicillin to the broth to ensure that the plasmid DNA would be maintained by the transformed cells. Overnight the cells have grown to high density and the plasmid DNA has undergone many replications. However since you started with a single colony of bacteria and that colony grew from a single transformed cell, all the copies of the plasmid DNA in your overnight culture should be identical (“clones” of one another). To isolate the plasmid DNA from an non-inducible bacterial strain, you will perform what is commonly called a “mini-prep”. This term distinguishes the procedure from a “maxi-” or “large scale-prep” which involves a larger volume of cells and additional steps of purification. The overall goal of each “prep” is the same--to separate the plasmid DNA from the chromosomal DNA so that certain genes on the plasmid DNA can be studied further.
TO DO TODAY:
To isolate plasmid DNA from an overnight culture of cells, the media is removed from the cells by centrifugation. The cells are resuspended in “Solution I” which contains Tris to buffer the cells and EDTA to bind divalent cations in the lipid bilayer, thereby weakening the cell envelope. Upon the addition of “Solution II,” the chromosomal DNA and the plasmid DNA are denatured by the sodium hydroxide, and the cellular proteins and lipids are dissolved by the detergent, sodium dodecyl sulfate (SDS). The pH of the solution is returned to neutral by the potassium acetate in “Solution III.” At neutral pH the SDS precipitates from solution, carrying with it the dissolved proteins and lipids. In addition, the DNA strands renature at neutral pH. The chromosomal DNA, which is much longer than the plasmid DNA, renatures as a tangle that gets trapped in the SDS precipitate. The plasmid DNA renatures normally and stays in a water based solution. In this way the plasmid DNA is separated from the chromosomal DNA, the proteins and the lipids of the cell. Plasmid DNA can be precipitated out of solution in absolute ethanol and then put back into solution (in an appropriate concentration) in water. The ingredients and concentrations of a stock solution of all reagents (such as Solutions I, II, and III) can be found in the Media Recipes section of this section of the wiki.
Today you will transform the isolated plasmid DNA, pL4440, into an IPTG inducable strain of E.coli, HT115(DE3), and spread the transformed bacteria onto LB agar media supplemented with both tetracyclin (resistance confered by a gene expressed by the bacteria) and ampicillin (resistance confered by a gene expressed from the plasmid).
Protocols
Part 1: Plasmid DNA Isolation:
Only one of your two overnight cultures should appear cloudy with bacterial growth. If your control is cloudy, please inform your instructor.
- To start the plasmid DNA isolation, label 2 microfuge tubes with whatever name you have selected for your plasmid, plus your initials and team color.
- Check to be sure that your cells have not settled. If they have, mix the cultures but do NOT invert them because they will leak. Then pour some of the overnight culture so that both microfuge tubes are almost full. If you are nervous about pouring the bacteria, you can pipet 750 microliters into each tube twice, so that there is a total of 1.5 ml in each tube. The exact volume doesn’t matter but the tubes should be quite full when you close the cap.
- Place the tubes in the room temperature microfuge so that the hinges of each cap is facing out. Paying attention to this small detail will help you know where in the tube to find your pellets. While it is not essential to do this in this step, it is a good habit to get into since sometimes pellets are very small and hard to see. Be sure your tubes are balanced, then spin the tubes for two minutes at 8,000 rpm. Check the rcf speed of the centrifuge while it is spinning (by hitting the toggle that changes the readout) and record the rcf’s in your lab notebook. Relative centrifugal force (rcf) is what should be used in M&M to describe your speed since it is universal and rpm is rotor and centrifuge dependent.
- If the supernatant is clear, pour it into the waste beaker that is on your bench, then flick the tube with the cap open to remove the last few drops of liquid. The cell pellet will not fall out. If the supernatant is not clear, recentrifuge until it is.
- Resuspend your cell pellets COMPLETELY in 100 microliters of solution I. Pipet up and down until ALL of the cells are uniformly suspended in Solution I. Be sure to change tips between samples. Leave the cells at room temperature as you prepare solution II.
- To make solution II, mix 500 microliters of 2% SDS with 500 microliters of 0.4M NaOH in a microfuge tube. Close the cap and invert the tube several times to mix the contents. Add 200 microliters of solution II to each miniprep and invert the tubes five or six times to mix. In some cases the minipreps may appear to "clear" but don't worry if you don't see a big change in yours. Place the tubes on ice for five minutes.
- Add 150 microliters of solution III to each miniprep and immediately vortex each tube for 10 seconds with your vortex set at the highest setting. White clumps should appear in the solution after you vortex it. Place the tubes in the room temperature microfuge and spin for 3 minutes at the HIGHEST speed.
- While the minipreps are spinning, label another set of microfuge tubes with a name you have chosen for your plasmid and your initials or team color. The new tubes can be left at room temperature.
- A white pellet should be visible when you remove your minipreps from the microfuge. Use your P1000 to transfer 400 microliters of each supernatant to the appropriate clean microfuge tube. It's OK to leave some of the supernatant behind. Try to avoid transferring the white pellet.
- Add 1 ml of room temperature 100% ethanol to each new tube. The tubes will be quite full. Close the caps and invert the tubes at least five times to thoroughly mix the contents.
- Spin the tubes in the room temperature microfuge for 2 minutes at HIGHEST speed. It is important to orient your tubes so that the hinges are up this time, as the pellets are expected to be barely visible.
- Pour off the supernatants into the waste beaker on your bench then flick out the last few drops. The tubes do not have to be completely dry.
- Carefully, wash the pellets with 500 microliters of 80% ethanol. (You may need to make a few mls of 80% ETOH from the 100% ethanol stock provided). When you wash the pellets, add the 80% ethanol slowly and carefully, so that the liquid flows down the side of the tube away from the pellet. After the ethanol has been dispensed, immediately remove it with the same tip, making sure to keep the tip on the side of the tube that doesn't have your pellet. Some liquid will remain in the tube, and it can and must be removed using your P200, set to 100 microliters. Try to remove as much of the ethanol as you can (without removing your pellet!). The pelleted DNA will have to be completely dry before you add water or your DNA will not go into solution.
- After all the DNA pellets have been washed, put your tubes with their caps open in the SpeedVac that is at the back of the lab. If you were able to remove most of the ethanol with the P200, then after two or three minutes the last drops of ethanol will have evaporated and the pellet will be barely visible. If the DNA pellet is invisible please see your instructor to determine how much water to use in the next step.
- If you have a visible pellet of DNA, add 30 microliters of sterile water to each tube and vortex each tube extensively--- a full two minutes. The pelleted plasmid DNA must dissolve completely in the water. The liquid can be brought back to the bottom of the tubes by spinning for a few seconds in the microfuge. You can then combine the dissolved plasmid DNA into one of the two tubes so that you have approximately 60 microliters of plasmid DNA as one aliquot. Store the DNA on ice until you are ready to use part of it and then give the rest to your instructor to freeze. DO NOT DISCARD ANY OF YOUR PLASMID ISOLATE!
Part 2: Transformation of isolated plasmid DNA into HT115(DE3):
The HT115(DE3) bacterial cells are on the instructor’s bench You will transform some of your plasmid DNA into this strain. The cells are very fragile, so treat them gently.
A few weeks ago, the prep staff made a lot of competent HT115(DE3) cells using the Inoue Method Media:Inoue bacterial transformation.doc. Reference: Inoue H., Nojima H., and Okayama H. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96: 23-28. The cells were made competent to take up free plasmid DNA by this treatment. This treatment makes the cell wall and membrane more permeable and our transformation efficiency much greater but it weakens the cells; therefore you must keep them cold and mix them gently throughout the transformation (no vortexing!).
- Label the top or the side of the tube with HT115(DE3), pL4440 (your gene name), and your initials or team color.
- Start the transformations by pipetting 50 microliters of well mixed (gently!) chemically competent cells to the labeled microfuge tube prepared in the the last step.
- Add 3 microliters of your plasmid DNA to the tube. Pipet up and down once to mix the DNA and the cells. Close the cap and let the transformation mixture sit on ice for 10 minutes.
- Heat shock by incubating the transformation mix at 42°C for 90 seconds, exactly. This step must be timed exactly. Remove the tubes at the end of 90 seconds to your ice bucket while you get your LB ready.
- Pour 3 mls LB from the stock bottle into a clean and sterile 15 ml conical tube. By doing this, you will minimize the amount of LB that will be contaminated if you accidentally touch the media with something that is not sterile. Contaminated media looks cloudy, so be sure to swirl and examine the stock bottle of LB to make sure it is not contaminated.
- Add 500 microliters of the LB in your conical tube to the transformation mix. When pipetting the media, remember to release your thumb on your micropipet slowly, to avoid splashing the liquid on the end of the barrel. The barrel is not sterile and if you see the liquid touch it, then discard the media in the waste beaker and try again with a new tip.
- Once you have added the media, close the cap and invert the tube once or twice to mix the contents. Incubate at 37°C for 45 minutes.
- While the plasmid DNA is being taken up by the competent cells and the new genes provided by the plasmid are being expressed by the bacteria, label one LB + amp + tet agar plate. Label the bottom of the plate with the strain's identity (HT115(DE3)), the plasmid used (your unique plasmid name), the date, your initials and team color. You must label the bottom of the plate since the tops are easily switched. Put this plate in the hood with the blower on and with the lid ajar to dry the surface of the agar for about 10 minutes or until the surface looks dry but is not badly dehydrated.
- Once the transformation mix has incubated at 37°C for 45 minutes, invert it to mix the contents and pipet 100 microliters of transformed cells onto the center of that slightly dehydrated LB plate prepared in the previous step. Pour 5-10 glass beads onto the plate. Put the lid back on and gently swirl the bead all over the plate to spread the transformed bacteria around. When you are done - pour the beads into the beaker on the bench.
- Replace the lid and leave the agar plate undisturbed for a few minutes.
- Once it has dried enough that the surface doesn’t appear wet, invert the plate and incubate them at 37°C for 24-48 hours. The plate should be incubated with the agar side up so that condensation will not drip onto the surface of the agar and smear the colonies that will be growing there.
- Save the remaining transformation mix for 24 hours or until we are sure that there is at least one colony growing on each of your plate.
What would it mean if you had no colonies on your plate? Normally, you would expect to have around 100 pale color colonies on each plate. If you have at least one well isolated colony on the plate, you’re all set. After the 24 hour growth period the plate should be placed in the rack in the refrigerator labeled with your lab day. You will use a single colony from the plate to make an overnight broth culture on the day before next lab. If you have no colonies on one or more of your plates, please notify your instructor right away.
Before leaving lab today, give the rest of your isolated plasmid DNA to your instructor in a labeled microfuge tube. Make sure your tube is labeled with your name, lab day, plasmid name and color coded with a piece of tape in your team color.
RNAi Schedule of Experiments
RNAi General Information
Media Recipes
Lab 4: Picking your gene to RNAi
Lab 6: Induction of RNAi plasmid and C. elegans feeding
Lab 7: Single Worm PCR and Agarose Gel Electrophoresis
Lab 8: PCR Reaction Cleanup and Sequencing
