BISC220/S14: Mod 1 Lab 3

Purifying and Handling Proteins
Extraction:
The first step in the purification of a specific intracellular protein is extraction from the cells. Bacterial cells can be broken and their enzymes extracted or solubilized by a variety of techniques that may involve mechanical methods (grinding, etc.) or chemical lysis of the cells. The objective is to release the desired enzyme from the cells as gently as possible to retain activity of the enzyme.
Isolation and Purification:
There are several methodologies that can be employed for the purification of enzymes, many of which are discussed in your text. These include:
- differential solubility
- ion exchange chromatography
- affinity chromatography
- molecular sieve techniques
- density gradients
- electrophoresis
- electrofocusing
Most of these methods rely on differences in either the net charge or molecular weights of the proteins. Generally cruder, less time consuming methods are used in the initial processing of cellular extracts. This is necessary not only because of the large quantities of protein to be processed but also because of the complexity of the protein mixture.
One classic method for the partial purification of enzymes from crude extracts is based on the differential solubility of proteins. Proteins remain dissolved in solution because of their charged surface residues (amino acid side chains) which interact with the molecules of the solvent (water). If such interactions are prevented, the protein molecules will interact principally with one another forming huge aggregates that precipitate out of solution. A common method of enzyme precipitation is by the addition of inorganic salts such as ammonium sulfate.
Stabilization:
When working with enzymes, it is important to provide environmental conditions that will reduce the amount of enzyme denaturation that occurs during purification procedures. Denaturation results in loss of enzymatic activity. The following are some of the conditions that must be controlled:
- pH: Enzymes have multiple charges on their surfaces, which must be preserved to maintain the native 3-D structure and hence enzymatic activity. Buffers are used to maintain the enzyme solutions at a desired pH. The buffer must be in the appropriate concentration, have the correct pKa, and must not adversely affect the protein. See your general chemistry text for a review of buffers and other inorganic chemistry terms.
- Ionic Strength : The ionic strength of the buffer solution is usually critical. Many enzymes require a medium that has a greater ionic strength than provided by the buffer alone. Inorganic salts such as potassium chloride or sodium chloride, which increase the ionic strength, are commonly added to increase the stability of enzymes. Adding sucrose or glycerol to the buffer can often stabilize enzymes requiring a more hydrophobic environment. We will be using glycerol to stabilize β-galactosidase during storage at -80°C.
- Protection Against Sulfhydryl Oxidation: Enzymes may contain many sulfhydryl groups (SH). One or more may be required for the activity of an enzyme. If these sulfhydryl groups become oxidized, they form intra- or intermolecular disulfide bonds. If necessary, the most effective method of retarding such oxidation is the addition of a reducing agent to the buffer. Dithiothreitol (DTT) and β-mercaptoethanol (βME) are among the most common and effective reducing agents. β-galactosidase seems to be most stable in a reducing environment, so often β-mercaptoethanol is included in the solutions used to extract and assay the enzyme.
- Protection Against Heavy Metals: In addition to oxidation, sulfhydryl groups may react with heavy metal ions such as lead, iron or copper. Principal sources of metal ions are the reagents used to make up buffers, substrates, and water itself. Deionized or distilled water is used to make up reagents and a chelating agent such as EDTA (ethylenediaminetetraacetic acid) can also be added.
- Temperature: A rule of thumb is to keep enzymes as close to 0-4°C as possible during purification procedures. A few enzymes exist, however, that denature or inactivate at cold temperatures.
Basic Rules for Handling Enzymes
The following information is taken from a popular primer now published by Roche Pharmaceuticals, but first published in the December 1985 issue of BMBiochemica. by Boehringer Mannheim Biochemicals.
- For best stability, enzymes should be stored in their original commercial form (lyophilized, ammonium sulfate suspension, etc.), undiluted and at the appropriate, specified temperature.
- For enzyme solutions and assay buffers, use the highest purity water available. Glass distilled water is best. Deionized water, especially if passed through an old filter or a reverse osmosis device, may contain traces of organic contaminants which inhibit enzymes.
- Enzymes should be handled in the cold (0-4°C) at all times. Dilute for use with ice-cold buffer or distilled water. While using an enzyme solution or suspension at the bench, keep it in on ice.
- Dilute enzyme solutions are generally unstable. The amount of enzyme required for the experiment should be diluted within 1-2 hours of use. Enzymes should not be diluted for long-term storage.
- Enzymes, especially those that have been diluted, should be periodically checked for activity to ensure that any loss in activity is taken into account when designing an experimental protocol. Expiration dates on vials only refer to enzymes stored in the original form at the correct temperature.
- Do not shake crystalline suspensions (e.g. ammonium sulfate suspensions) since oxygen tends to denature the enzyme. The material should be resuspended with gentle swirling or by rolling the bottle on the lab bench. Once the enzyme crystals have been uniformly resuspended, remove the amount needed with a pipette. In many cases, the enzyme crystals may be used directly in the assay procedure.
- Do not freeze crystalline suspensions. Freezing and thawing in the presence of high salt concentrations causes denaturation and loss of activity.
- Vials containing lyophilized enzymes (as well as cofactors such as NADH and NADPH) should be warmed to room temperature before opening. This prevents condensation of moisture onto the powder, which can cause loss of activity or degradation. If the reagent is hygroscopic, mishandling may ruin the entire vial.
- Avoid repeated freeze-thawing of dilute enzymes and lyophilizates in solution. Store in small aliquots. Thaw one portion at a time and store that portion once thawed at 4°C. The stability of individual enzymes may vary greatly and often should be determined empirically under your exact conditions.
- Detergents and preservatives should be used with caution, since they may affect enzyme activity. Sodium azide, for example, inhibits many enzymes which contain heme groups (e.g. peroxidase). Detergents added at concentrations above their critical concentration form micelles which may entrap and/or denature the enzyme.
- Enzymes should be handled carefully. To avoid contamination of any kind, use a fresh pipette tip for each aliquot that is removed from the parent vial. Never return unused material to the parent vial. Wear gloves to prevent contaminating the enzyme with proteases, DNAses, RNAses, and inhibitors often found on fingertips. Never pipette by mouth.
- Adjust the pH of the enzyme buffer at the temperature at which it will be used. Many common buffers (Tris, glycylglycine, Bes, Aces, Tes, Bicine, Hepes) change rapidly as the temperature changes. For instance, Tris buffer decreases 0.3 units of pH for EVERY 10°C rise in temperature. A solution of Tris, adjusted to pH7.5 at +25°C will have a pH of 8.1 at 4°C or 7.2 at 37°C. The change in pH per 10°C temperature change for other buffers is: Aces, -0.20; Bes, -0.16 ; Bicine,-0.18; glyclyglycine, -0.28; Hepes,-0.14; Tes, -0.20 [Good, N.E., Winget, G.D., Winter, W., Connolly, T.N., Izawa, S., and Singh, R.M. (1966). Hydrogen ion buffers for biological research. Biochemistry 5, 467-77.]
- The absorbance at 280nm is widely used to quickly determine the protein concentration of an enzyme solution. However, this absorbance is due to the presence of tyrosine and tryptophan in the protein. If an enzyme (e.g. superoxide dismutase) contains low amounts of these two amino acids it will not absorb significantly at 280nm.
Resources:
Detailed information is available on many enzymes. The following are excellent resources.
Methods in Enzymology, published by Academic Press, Editors in chief: Sidney P Colowick and Nathan O. Kaplan. There are more than 165 volumes in this series, covering an extensive range of topics.
The Enzymes, 3rd edition, edited by Paul D. Boyer. An excellent, broad series which focuses more on the properties of the enzyme and less on methodology than Methods in Enzymology.
Methods in Enzymatic Analysis, 3rd edition. Editor-in-chief: Hans U. Bergmeyer, published in Verlag Chemie. In-depth discussions of techniques of analysis that use enzymes or that assay enzymatic activity.
Affinity Chromatography
The use of metal chelate affinity chromatography for protein purification was first reported by Porath and colleagues in 1975. This landmark report applied the knowledge that histidine and cysteine form rather stable complexes with some cations such as zinc and copper ions. We now know that the amino acid tryptophan shares this characteristic with histidine and cysteine and that some proteins have specific binding sites for these metals. Porath et al. (1975) devised a method to tightly bind metal ions to a solid matrix such as agarose beads. The matrix resides in a column through which protein solutions are passed. Some proteins in these solutions bind to the matrix but can be specifically eluted by a low pH wash solution. Later work (Porath and Olin, 1983; Kagedal, 1998) showed that it is possible to release proteins from such columns using a strong soluble chelator such as EDTA or by chemically competing with the binding. For example, histidine binding can be disrupted by including imidazole in the protein elution buffer. The chemical structure of imidazole is similar to the ring structure of histidine, therefore it competes for the binding site on the cation matrix.
In today’s lab, we are using agarose beads chelated with nickel to fractionate the 6xHis-tagged β-galactosidase. Nickel effectively binds histidine, and the combination of 6 histidine residues in a row at the amino terminus of β-galactosidase should lead to rather tight binding. After washing the nickel chelated agarose to remove any unbound or loosely bound protein, we will use a buffer containing 200 mM imidazole to release the 6xHis-tagged enzyme from the nickel-chelated agarose beads.
Affinity Column Purification Protocol
In this laboratory session you will purify β-galactosidase from the cell pellet of the genetically modified E. coli you induced last week to overexpress this enzyme.
Using B-Per 6xHis Fusion Protein Purification Kit©, product #78100 by Pierce, Inc.
Microsoft Word File: Purification of 6xHis-tagged β-gal
- Defrost the frozen pellet of E.coli BL21(pET-14b) cells prepared in the previous lab session and add 10.0 mL of B-Per Protein Extraction Reagent™, a mild anionic detergent (the exact ingredients of which are proprietary), to the centrifuge bottle. Add 20μL of DNAase (to achieve a concentration of 1 unit/mL from the 5000units/ml stock) to the B-Per and cells. Resuspend the cells by vortex mixing and pipetting up and down until the pellet is off the side of the bottle and all clumps are gone. Try to avoid bubbles. This should take about 5 minutes total, of alternately mixing and letting the mixture sit in your ice bucket. Your goal is to dissolve the cell walls and membranes which will lyse the cells and allow degradation of the DNA. Ask your instructor to check your suspension before proceeding to step 2. If you have obvious clumps, the cells are less likely to be lysed sufficiently.
- Shake the suspension gently for 10 minutes at room temperature using a platform shaker. During this time the lysed cells will release their contents and soluble bacterial proteins will be dissolved in the lysate. Be sure to break up any obvious clumps with gentle vortexing.
- Carefully pour the entire suspension into a Corex tube fitted with a rubber adapter and centrifuge for 15 minutes at 10,500 rpm in the Sorval refrigerated centrifuge at 4°C using an SS-34 rotor. You will need to make a balance tube with water. Be sure and record in your lab notebook the speed as g force (use the chart near the centrifuges to make the conversion).
- Pour the supernatant only into a 15 mL graduated conical tube. This solution of dissolved proteins in B-Per reagent is your cell-free or Crude Extract (CE). The pellet is bacterial cell debris that can be dumped into your waste container. The Corex tubes are NOT disposable. Please rinse them out and leave them on your bench or return them to your instructor.
- Determine and record the total volume of the crude extract (CE) using the volume markings on the tube. Pipet 600 µL of CE into a microcentrifuge tube labeled with your team color, lab day, initials and "CE" or “Crude Extract.” Store the aliquot on ice. At the end of lab today, you will give what remains of this aliquot to your instructor to freeze for use next time.
- Add 1000µL (1mL) of the Nickel-Chelated Agarose to the volume of CE remaining in the 15mL conical tube. It is essential that the agarose suspension be properly mixed before removing an aliquot, so mix it well by swirling or gentle vortexing just before removing it from the stock.
- Shake the Agarose-CE mixture gently for 10 minutes at room temperature on the platform shaker. Make sure the cap is secured tightly. Centrifuge for 5 minutes at top speed in a clinical, benchtop centrifuge.
- Using a Pasteur pipette, carefully remove and discard the supernatant solution into the provided waste container. Do not try to pour off the supernatant! The 6xHis-tagged proteins should now be bound to the nickel-chelated agarose beads at the bottom of the tube.
- Add 3.0 mL of Wash Buffer #1® to the agarose beads in the conical tube, and resuspend using a pipette. Do not vortex. Gently shake the suspension at room temperature for 5 minutes on the platform shaker. Centrifuge for 3 minutes in a clinical, benchtop centrifuge. Carefully remove supernatant and discard.
- Add 3.0 mL of Wash Buffer #2® to the centrifuge tube and resuspend the agarose using a pipette. Do not vortex. Gently shake the suspension at room temperature for 5 minutes on the platform shaker. Centrifuge for 3 minutes in a clinical, benchtop centrifuge. Carefully remove supernatant and discard.
- Add 250µL of Wash Buffer #2® to the agarose, resuspend completely and transfer all of this suspension from the centrifuge tube to a B-PERTM Spin Column that has been placed into one of the special collection tubes provided. To insure that the agarose beads don’t get stuck in the pipet tip of your P1000, cut about a 1/4 inch off the end of it with a single edged razor blade. Centrifuge for 2 minutes at maximum speed in a microcentrifuge. Discard the collection tube with the flow-through.
- Place the spin column in a new collection tube and add 500µL of Elution Buffer containing imidazole. Mix the agarose and the buffer in the spin column using the NOT DISPOSABLE plastic mixer provided by your instructor. Incubate the spin column at room temperature for 5 minutes. During this time, most of the 6x His-tagged protein will be eluted from the nickel-chelated agarose into the buffer. After the 5 minute incubation period, recover the protein by centrifuging for 2 minutes at top speed in a microcentrifuge. Your purified fraction will be the flow-through in the collection tube. If you have considerably less than 500µL, respin the column to collect more volume.
- Take the spin column out of the collection tube and discard the spin column. Do not discard the material in the collection tube. Using a Pasteur pipette, transfer all of the contents containing the purified β-galactosidase to a new microfuge tube with volume markings. Record the volume in your notebook! Label the tube to indicate that this is the purified β-galactosidase fraction (e.g., "PF β-gal"), and save it in your ice bucket.
DO NOT DISCARD ANY OF THE CRUDE EXTRACT OR PURIFIED FRACTION!
You are now ready to assay both the Crude Extract and Purified Fraction for total protein, using a modified Bradford Dye Assay (Bio-Rad assay). You will later assay both CE and PE for β-galactosidase activity (enzyme function) using a different assay.
Modified Bradford Dye Assay (Bio-Rad™ Assay) for Total Protein
We will now determine the total protein concentration in our CE and PF fractions in order to load each lane of our gel with equal amounts of protein. Loading lanes equally is the ONLY way we can make comparisons between lanes.
Microsoft Word File: Media:Modified Bradford Dye Assay Protocol.doc
Testing the quality of the purification:
The total protein assay of choice is a spectrophotometric method known as the modified Bradford dye assay. It is also sometimes called the “Bio-Rad™ Assay" because the assay reagent was purchased from a company named Bio-Rad. It is a dye-binding assay based on a differential color change of Coomassie Brilliant Blue in response to various concentrations of protein. The absorbance maximum for an acidic solution of Coomassie Brilliant Blue G-250 shifts from 465 nm to 595 nm when binding to protein occurs. In order to determine protein concentration in unknowns (CE and PF) we must compare their absorbances to the absorbance of known concentrations of protein that have been used to create a standard curve. Please note that this assay does not measure only our protein of interest, β-galactosidase, but instead, measures the concentration of all the proteins, including β-galactosidase, that are present in our starting and our purified material. We measure total protein for two reasons: we need to use the total protein concentration determined by this assay in order to determine the concentration of β-galactosidase in our specific activity assay and because knowing the total protein concentration in our starting vs. purified material helps us determine how much contaminating protein we removed during our purification.
Dilution Preparation for the Standard Curve
Prepare a set of 6 microfuge tubes. Dilute a stock solution of bovine serum albumin (BSA) with a concentration of 1mg/ml to yield 200 microliters of BSA in concentrations of: 0.1, 0.2, 0.4, 0.6, 0.8 and 1 mg/ml.
Table 2: Dilution of Stock BSA to make protein standards for total protein assay
| Tube # | BSA Concentration | Z Buffer in μL | Stock BSA |
|---|---|---|---|
| 1 | |||
| 2 | |||
| 3 | |||
| 4 | |||
| 5 | |||
| 6 |
The diluent will be Z-buffer (60mM Na2HPO4, 60mM NaH2PO4, 1mM MgSO4, 0.27% beta-mercaptoethanol) and the final volume needed is 200 μl. The formula C1 x V1 = C2 x V2 should help you. Check with your instructor to confirm your dilution strategy in the table above before making these working concentrations of the standards. Remember to add the smaller volume to the larger volume.
Dilution Preparation of Unknowns:
Dilute your CE and PF 1:5 in Z buffer to yield a final volume of 300 μl of each.
We are trying to determine the concentration of protein in each but since we don’t know it at this point we will have to use a different dilution formula.
sV x df = ftV where sV is starting volume (the unknown); df is the dilution factor (5); ftV is the final total Volume (300μl). Check your dilution strategy with your instructor before making the dilutions.
Amount of Z-buffer required = ?
Amount of CE or PF required = ?
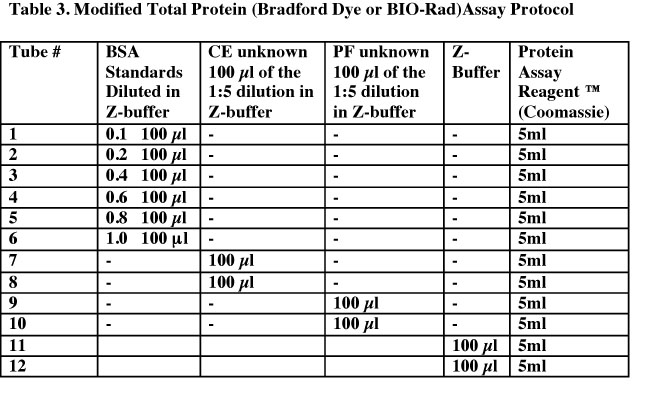
Total Protein Assay Protocol:
| Tube # | BSA Standard Diluted in Z-Buffer | CE (1:5 dilution in Z-Buffer) | PF (1:5 dilution in Z-Buffer) | Z-Buffer | Protein Assay Reagent™ |
|---|---|---|---|---|---|
| 1 | 0.1 mg/ml (100 ul) | - | - | - | 5 ml |
| 2 | 0.2 mg/ml (100 ul) | - | - | - | 5 ml |
| 3 | 0.4 mg/ml | - | - | - | 5 ml |
| 4 | 0.6 mg/ml | - | - | - | 5 ml |
| 5 | 0.8 mg/ml | - | - | - | 5 ml |
| 6 | 1.0 mg/ml | - | - | - | 5 ml |
| 7 | - | 100 ul | - | - | 5 ml |
| 8 | - | 100 ul | - | - | 5 ml |
| 9 | - | - | 100 ul | - | 5 ml |
| 10 | - | - | 100 ul | - | 5 ml |
| 11 | - | - | - | 100 ul | 5 ml |
- Label a set of 12 glass tubes. See Table 3 below for the contents of each tube. Put the Protein Assay Reagent into all tubes last. Cover the tubes with parafilm and mix well by vortexing.
- Let the reactions and blanks incubate at RT for a minimum of 15 minutes. The reaction is stable for one hour.
- You can start making your dilutions or set up your assay tubes for the specific activity assay while you are waiting for the total protein assay to maximize its absorbance shift.
- After maximum color development (at least 15 minutes but not more than one hour) mix all 12 tubes again and transfer enough of the contents of each assay tube or blanks to a set of labeled 1.5mm plastic cuvettes so that the cuvettes are at least 2/3 to 3/4 full. Check to be sure that there is no precipitate interfering with light passage. If so, let the precipitate settle to the bottom before reading absorbance.
- Follow the directions in the appendix for reading absorbance using either the Hitachi or Cary 50 spectrophotometers. Set the wavelength to 595nm.
- Blank the spectrophotometer using the reagent blank(s) (tubes 10, and or11).
- Read A595 for all tubes and record in your lab notebook.

Tubes 1-6 (the diluted BSA standards) contain known concentrations of protein. You will use those known concentrations and absorbances to create a standard curve and linear regression line using Excel (see Linear Regression Instructions). The creation of a standard curve will allow calculation of the protein concentration in mg/ml from the absorbance of the diluted CE and PF. Make sure your R2 value for the line is at least .95. The CE and PF have been assayed in duplicate, therefore, the concentrations can be averaged if the replicates are within 10% of each other. Record the total protein concentration for both the CE and the PF in your lab notebook. Remember that the true concentration will be 5 times the value obtained from your linear regression formula since the assay used a 1:5 dilution.
Polyacrylamide Gel Electrophoresis
Another way to determine the effectiveness of our purification technique is to perform sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis on the CE and PF. SDS, an anionic detergent, is used to both denature the proteins and to give them an overall negative charge. The electrophoresis loading and running buffers also contain SDS. This negative charge allows the proteins to run toward the anode (positive electrode). The larger molecules are retarded by the acrylamide, which acts like a sieve, so they move more slowly through the gel than the smaller molecules. By running a sample of known molecular weights in a lane adjacent to the extracts, it is possible, by comparison, to determine the molecular weights of the bands of unknown proteins in each of the extracts. In addition to SDS, a denaturing and loading solution called Laemmli buffer contains:
- 10% glycerol, which makes the solution dense enough to fall through the running buffer into the wells of the gel, so the sample doesn't float away;
- 5% β−mercaptoethanol to break sulfide bonds and to keep disulfide bonds from reforming between the denatured proteins
- 2% SDS Sodium dodecyl sulfate,
- a trace amount of tracking dye, bromphenol blue, which will run ahead of the proteins, forming a visible front as the samples run during electrophoresis.
- in the solvent 0.0625 M Tris buffer pH 6.7
The gels are placed in the tank, which is half filled with cold running buffer. The top chamber is filled and checked for leaks, which can also cause distortion of the current. The current runs from the top chamber through the gel to the lower chamber, which is connected to the anode (red electrode). The negatively charged proteins will migrate toward the anode according to their molecular weights with the smaller proteins migrating farther down the gel than the larger proteins.
Protocol
Microsoft Word File: Media:Gel Electrophoresis Protocol GelCode.doc
For SDS-PAGE, you will load 10 µl of each of your 4 samples (CE, PF, pre-IPTG cells, and post-IPTG cells) into lanes of a 4-15% gradient polyacrylamide gel. The crude extract and purified fraction should each contain 5 µg of protein.
- Make dilutions in Z buffer of your crude extract (CE) and your purified fraction (PF) to end up with 15 μg of protein in 15 μl of volume (1µg/µl concentration). You will have to look up the original protein concentration that you calculated for these samples. Remember that the sample of PF that you will use today has no added glycerol. **If you had less than 1 mg/ml of PF then you cannot make a dilution! See your instructor.
- Add an equal volume (15 μl) of Laemmli buffer, also called sample buffer. You will load 10 μl of this mixture onto the gel. The sample loaded contains 5 micrograms of protein. Your Instructor will demonstrate the loading procedure.
- In Lab I you saved some of the E. coli cells harvested before and after the β-galactosidase induction step with IPTG. To each of the 2 cell pellets add 75 μl of Laemmli sample buffer and resuspend the cells completely in the buffer.
- Boil all four samples for 5 minutes. Do not boil the molecular weight standard, but do boil the commercial β-galactosidase positive control along with your samples.
- Each electrophoresis chamber holds 2 gels, so 2 groups will run their gels together. Each group will load 6 lanes in one 4-15% gradient polyacrylamide gel according to the template below. Load 10µl of sample into each lane of your gel including a β-galactosidase positive control and the molecular weight standards. Be sure and take a copy of the key to the standards.
- Your instructor will connect the power source and set it to 200 volts. If no current reading (amps) appears, there is probably a leak in the upper chamber and extra running buffer has to be added to the upper chamber.
- After approximately 45 minutes, the tracking dye should have migrated to the bottom of the gel. TURN THE POWER OFF. Remove the gels from the reservoir. Your instructor will demonstrate how to remove the gel from between the plastic plates. Wear gloves when handling the gels because acrylamide is toxic.
- Place the gel in your plastic box and add enough deionized H2O to cover your gel and have it float freely. Incubate at room temperature with gentle rocking for approximately 5 minutes - repeat for a total of 3 times.
- Pour off the water and add enough GelCode Blue Stain Reagent to cover your gel and have it float freely. Return the gel to the shaker and stain for at least 30 minutes.
- To destain the gel add enough deionized H2O to cover your gel and have it float freely. Incubate at room temperature with gentle rocking for approximately 5 minutes - repeat for a total of 3 times.
- Photograph your gels using white light trans-illumination. Gel images will be posted to the conference as jpg files for incorporation into your lab reports.
| Lane # | Sample Name |
|---|---|
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 |
Preserving the Enzymatic Activity of the CE and PF
Glycerol acts to preserve enzymatic activity while the samples are frozen. We will be using these aliquots of crude extract and purified fraction to study the enzyme kinetics of β-galactosidase in Lab 4.
- Determine the remaining volume of the purified fraction and add an amount of 70% glycerol equal to 1/2 the volume of that fraction. Mix well by inversion. For example, if your purified fraction has a volume of 250 µl, you would add 125 µl of 70% glycerol solution it.
- Pipette 750 ul of Crude Extract (CE) into a clean microfuge tube. Label it CE. Add 375 μL of 70% glycerol to this fraction. Mix well by inversion.
- Give both labeled tubes to your instructor to save for next week.
Assignment
- Using measurements obtained from your gels plot the log10 of the molecular weights of the standard proteins (y-axis) versus mobility using the Instructions for Determining Molecular Weight.
- Using the standard curve generated in the previous step, calculate the molecular weight of the band(s) that you have identified as β-galactosidase. What is the calculated molecular weight?
- How well do your measurements agree with other published values for the molecular weight of β-galactosidase? Propose an explanation if your molecular weight differs significantly from the literature.
- Is the MW calculated above the same as that of the native β-galactosidase that you purified and assayed for enzymatic activity? Why/why not? How could you determine the answer to this question?
- Which of the bands on your gel exhibit the largest change in response to the addition of IPTG?
- Was β-galactosidase present before the addition of IPTG? How do you know?
- What other experiments could you do to prove that the band is β-galactosidase if your reader is unconvinced by the evidence of its migration at the same place as the standard?
References
The references cited here are on reserve for BISC 220 Cellular Physiology either electronically or in hard copy in the Science Library.
Background information on enzymes and lab techniques:
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002) Molecular Biology of the Cell 3rd ed. Garland Pub., New York.
Hawcroft DM (1997) Electrophoresis The Basics IRL Press, New York.
Kågedal, L. (1998) Immobilized Metal Ion Affinity Chromatography. In: Protein Purification (Janson, J. C. and Rydén, L., eds.), Wiley-VCH, New York, NY, pp. 311–342.
Lehninger AL, Nelson DL, Cox MM (1993) Principles of Biochemistry 2nd ed. Worth Pub, New York.
Porath J, Carlsson J, Olsson I, Belfrage G (1975) Metal chelate affinity chromatography, a new approach to protein fractionation. Nature 258: 598-599.
Porath J, Olin B (1983) Immobilized metal ion affinity adsoprtion and immobilized metal ion affinity chromatography of biomaterials. Serum protein affinities for gel-immobilized iron and nickel ions. Biochemistry 22: 1621-1630.
Williams G (1998) HyperCell CD-ROM. Garland Pub., New York.
Specific References for β-galactosidase:
Jacobson RH, Zhang X-J, DuBose RF, Matthews BW (1994) Three-dimensional Structure of β-galactosidase from E. Coli. Nature 369; 761-766.
Juers DH, Hakda S, Matthews B, Huber RE (2003) Structural basis for the altered activity of Gly 794 variants of Escherichia coli β-galactosidase. Biochemistry. 42: 13505-13511.
Juers DH, Heightman TD, Vasella A, McCarter JD, Mackenzie L, Withers SG, Matthews BW (2001) A structural view of the action of Escherichia coli (lacZ) β-galactosidase. Biochemistry. 40: 14781-94.
Roth NJ, Rob B, Huber RE (1998) His–357 of β-galactosidase (Escherichia coli) Interacts with the C3 Hydroxyl in the Transition State and Helps To Mediate Catalysis Biochemistry 37: 10099-10107.
Ring, M, Huber RE (1990) Multiple replacements establish the importance of tyrosine 503 in β-galactosidase (Escherichia coli). Arch Biochem Biophys 283: 342-350
