BISC219/F12: RNAi Lab 9

Lab 9: Series 3 - Induction of the Feeding Strain to Produce dsRNA and RNAi
To do on the day before the next lab:
You and your partner will return to the lab to make an overnight broth culture of your selected colony as described below. This process will create a sub-culture of many identical copies of the bacteria containing the plasmid carrying the construct to RNAi the gene that you want to study.
- Find your plate in the glass front refrigerator in a rack labeled with your lab day. Select a large, well isolated colony to start your overnight culture. Do not pick a small "satelite" colony as it may not be transformed with our plasmid.
- Begin by obtaining two tubes of LB broth (each will have 5 ml of broth) from the refrigerator in the back left hand corner of the room.
- Add 5 microliters of the 50mg/ml ampicillin stock (also found in the refrigerator with the broth) to each tube. Calculate the effective concentration of ampicillin that you have in your LB tube (remember V1 x C1= V2 x C2) and record that information in your lab notebook.
- Add 5 microliters of the 12.5mg/ml tetracycline stock (also found in the refrigerator with the broth). Calculate the effective concentration of tetracycline that you havein your LB broth tube. Record that info in your lab notebook.
- Gently swirl your LB +amp+tet broth to mix the contents.
- Label the two sterile glass culture tubes with tape in your team color. Label one with "pPD129.36 lsy-2" and your initials. Label the other with your initials only.
- Inoculate the broth with your bacteria by using a sterile disposable loop to scrape your candidate colony off the plate. Be sure not to touch the plate with the loop except on the desired colony and don’t pick up any satellite colonies! Gently swirl the loop in the LB+amp+tet broth - you should be able to see the colony come off the loop. (The second tube of broth labeled with just your initials is a control and should not be inoculated with bacteria as it is your control for contamination.) If you prefer to use a sterile toothpick rather than a loop, you may pick up the colony with the sterile toothpick and drop the toothpick into the broth culture. Note that the tip with the colony is in the broth and the contaminated part you touched with your fingers does not touch the sterile medium.
- Balance the 2 tubes across from each other on the rotating wheel in the 37C incubator at the front of the room when you come in the door. DO NOT USE THE ROOM TEMP WHEEL!
- Incubate these broth cultures at 37°C overnight. Do not forget to make sure the wheel is rotating when you leave!
To Do Today
Through reverse genetics we will deduce the function of a gene starting with its sequence and working back to its phenotype. There are many genes in the genome whose phenotype when mutated is lethal; therefore, it is very difficult to near impossible to tie function to a particular gene in the traditional forward genetics manner of creating random mutations, looking for phenotype changes, and then finding the defective gene responsible for that function.
In our reverse genetics study of an interesting C. elegans gene, two different strains of worms, wild-type and rrf-3 (RNAi enhanced), are fed bacteria expressing dsRNA specific to a particular worm gene. Ingesting dsRNA initiates cascade of events that leads to the destruction of the mRNA of the target gene. An altered phenotype in the progeny of RNAi-treated worms indicates what happens when the normal function of this gene is lost or significantly downregulated.
Double stranded RNA (dsRNA) can be introduced to the C. elegans cells in many different ways including: feeding, injection and soaking. Each of these methods has positives and negatives. We are using the feeding method - where we use genetically modified bacteria as dsRNA factories.
To begin to investigate the power of reverse genetics, you will need to grow your own induced bacteria to seed your plates for RNAi feeding.
Done on the night before this lab:
You and your partner made an overnight broth culture of your selected colony. This process created a sub-culture of many identical copies of the plasmid carrying the construct that will induce RNAi to silence or downregulate the gene that you want to study.
On the morning of lab:
Your instructor or the lab staff will come in early in the morning and sub-culture your bacterial overnight. The cells will be in stationary phase in the morning and successful induction requires log phase growth.
To create the subculture of bacteria your cultures will be diluted 1:5 (1 mL of culture into 4 ml of LB + amp). To make enough induced bacteria for your experiment, each group will have two induced cultures. These cultures will be allowed to grow for three hours and then induced to make lots of dsRNA by adding IPTG to the culture and letting it continue to incubate for a few hours so the cell is full of dsRNA. The IPTG will compete with the repressors on the lac o promoter and remove them and allow the gene for T7 RNA polymerase to be transcribed and then translated into the RNA polymerase protein. The T7 RNA polymerase then binds to the T7 promoters on the pPD129.36 plasmid and transcribes our C. elegans lsy-2DNA into RNA!
A simplified map of the C. elegans RNAi plasmid :
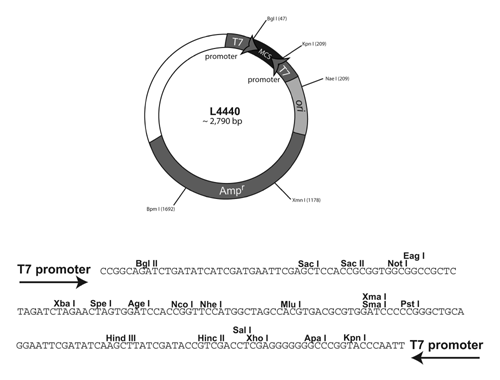
More specifically: The bacteria cells of strain HTll5(DE3) contain the T7 RNA polymerase gene (contained within a stable insertion of a modified lambda prophage λ DE3) under the control of lac operon regulatory elements. This allows expression of T7 polymerase to be controlled by isopropyl-β-D-thiogalactopyranoside (IPTG), a lactose analogue that induces expression of genes under the control of the lac operon o gene. When IPTG is added, the cells will begin to synthesize lots of T7 RNA polymerase. This T7 RNA polymerase can then bind to the T7 promoter sites on the plasmid and begin to synthesize RNA from both T7 RNA polymerase sites. Because the two single strands of RNA are complementary to each other they will form double stranded RNA within the bacterial cell. The IPTG induction allows us to "turn on" and express the plasmid gene of interest only when we want to and it allow us to make much higher levels of RNA for RNA interference than would be made without this induction.
Another useful thing about E. coli strain HT 115(DE3)is that this particular strain is deficient for the RNAaseIII enzyme that degrades double stranded RNA (dsRNA) in the bacterial cell. This allows for the accumulation of dsRNA in the cell and, thus, our ability to induce and RNAi effect! This E. coli strain carries a tetracyclin resistance gene so these cells can be selected on media containing tetracyclin, while the plasmid contains an ampicillin resistance gene that allows only transformed cells to grow on media containing ampicillin.
Our goal is to upregulate production of dsRNA of our worm gene of interest in pPD129.36 plasmids containing that gene in HT115(DE3) bacteria.
To induce your cultures, the lab specialist or your instructor:
- Added 5 μL of 0.5 M IPTG to 5ml of a log-phase sub-culture of your cells. What is the effective concentration of IPTG?
- Put your culture back in the 37°C incubator in the spinning wheel for approximately 3-4 hours.
- The bacteria worked hard expressing T7 RNA polymerase and transcribing complementary mRNA of C. elegans' lsy-2".
To do after induction is complete:
- Pipette 1.5 ml (use your P1000 to pipet 750μL twice) of your induced culture into 4 identical microfuge tubes.
- Spin your culture in a table top centrifuge for 5 minutes at 3000 rpm.
- Remove all of the supernatant.
- Resuspend the bacterial pellets of each tube in 100μL of LB - you are concentrating your bacteria.
- In the laminar flow hood in L304 or L301 (labs next door) pipet all your induced bacteria onto the center of 4 feeding plates that you have pre-labeled with an F. Be careful not to tilt or jostle your plates so that the bacteria stay in a circle in the center of each plate. These plates contain the same NGM Lite medium used in our mapping series, except that they have been supplemented with 0.4 mM IPTG, 50 μg/mL ampicillin.
- Allow the bacteria to be absorbed into the medium a bit before you try to move them. Being very careful not to "slosh" the medium around the plate, put your plates in the 37C incubator for ~3 hours.
- Obtain 2 control plates and label each of them with a C - these plates contain the same NGM lite medium described above and the bacterial strain on them are identical to your RNAi feeder strain, except that the pPD129.36 plasmid is only expressing RNA from the original multiple cloning site region (MCS) of vector - it lacks DNA specific to any worm genes.
- At the end of lab today, using a sterilized scalpel, cut 3 small (0.5 cm x 0.5 cm) squares from a starved wild type worm culture.
- Using the same scapel, scoop a square and place one square onto a pPD129.36 +lsy-2 feeding plate. Repeat to make a replicate.
- Place the third square onto a control plate.
- Flame your scalpel blade, cool it for a few seconds and cut 3 similar squares from a starved rrf-3 worm culture.
- Place one square onto each of two pPD129.36+lsy-2feeding plates and another square onto a control plate.
- Incubate all the worms for 7 days at 16°C
Preparation of the Chemotaxis Test Plates
Obtain from your instructor 8 unseeded salt plates. These will be your chemotaxis assay plates. With the lid on, draw the large unfilled and small filled circles on the bottom of the plate exactly as shown. Use the template your instructor will provide and make sure the distances are as the drawing below suggests:
 Image from Andersen et al. (2008)
Image from Andersen et al. (2008)
Once your plates are labeled wrap them with an elastic and a piece of your team tape and put them in the box provided by your instructor. The final steps to prepare these plates including the gradient will be described in the next lab.
