BISC219/F12: RNAi Lab 7

Lab 7: Series 3-Reverse Genetics: Investigating Gene Function & Regulation Using RNAi
In the age of genome sequencing we now know, or can make educated guesses about, the location of every gene in an organism's genome; however, this does not give us any information about the function of the gene product (protein) in the organism. We can use reverse genetic analysis to help us solve this puzzle. There are several tools in the reverse genetics toolbox: directed mutation (point mutations or deletions), overexpression using transgenes, and gene silencing using knockout organisms or double stranded RNA (RNAi). Only RNAi and overexpression have been perfected in C. elegans. Scientists still have not found a way to do in vivo homologous recombination in worms.
We are going to use RNAi as our tool to investigate gene function via reverse genetics. C. elegans is the first animal in which the process of RNAi was discovered. A similar system was identified in plants years earlier but, curiously, that groundbreaking discovery was largely ignored by the scientific community until it was noticed in animal models. We now know that RNA regulation in cells is a fundamental method of regulating gene expression in organisms ranging from microscopic C. elegans to humans. Many labs are now working non-stop to develop treatments for many "incurable" human diseases using RNAi.
In this investigation for Series 3 that you will begin today, each pair of students will be given E. coli bacteria that have been genetically manipulated to contain a plasmid that contains the C. elegans lsy-2 gene. The bacteria that contain this plasmid express C. elegans lsy-2 double stranded RNA. These bacteria will serve as food for our wild type C. elegans (worms with normal copies of lsy-2) and induce the RNA interference pathway in those worms. The result should be a '"knock down" in the amount of mRNA specific to the lsy-2 gene and, thus, a reduction in the amount of lsy-2 gene product. We hope to see the effect of this gene silencing in phenotypic differences between wild type worms that were RNAi induced and those that were not.
Calibration of Micropipettes
Before we begin our molecular genetic experiments, we will practice (AGAIN) properly handling and using the micropipettes.
- To calibrate your P1000, P200, and P20 micropipets, label 6 microfuge tubes (1-6) and weigh them. Record the weights in the table below.
- Following the table below, pipet the specified volumes into the pre-weighed microfuge tubes prepared above and then re-weigh them. Record all weights.
- Calculate the weight of the water in grams. 1000 microliters of water should weigh 1 gram at room temperature.
- If the water in any tube weighs more or less than 1 gram, ask your instructor for help. If your calibration is significantly off after several repeated attempts, your pipet (or your technique!) may need adjustment.
| Tube # | Tube Pre-Weight | Vol. in μL using P20 | Vol. in μL using P200 | Vol. in μL using P1000 | Weight of Tube + Water in grams | Weight of Water in grams |
|---|---|---|---|---|---|---|
For a Word™ format protocol: Media:Protocol for Micropipet Calibration.doc
Picking a Bacterial Colony
Your instructor will give each pair an LB+ampicillin plate on which TOP10 bacteria containing our pPD129.36 lsy-2 plasmid are selectively growing. Please pick two well isolated colonies, circle them, label one A and the other B and write your initials and group color on the plastic bottom (agar side) of the plate.
How can you be sure that your colonies contain the plasmid carrying the gene you are interested in studying? In theory, any colony of bacteria growing on your LB+amp plate should contain a plasmid because the gene for antibiotic resistance is not chromosomal, but instead expressed from your plasmid. Because only transformed bacteria are resistant to ampicillin, if we grow the bacteria on or in a medium containing ampicillin, those bacteria that did not take up plasmid DNA should not be able to reproduce to form colonies while those that express plasmid gene products and transfer the plasmid to their progeny will form colonies. The amp resistance gene on the plasmid encodes an enzyme called beta-lactamase. This enzyme is a secreted, soluble protein, which means that there may be smaller, non-transformed, "satellite" colonies around a true transformant. This happens because the ampicillin in the media is destroyed in the area immediately around the colony secreting the enzyme; therefore, there is no ampicillin in the area around the transformant and non-transformed cells can grow and divide enough to form smaller, satellite colonies. You must be careful to pick ONLY the bigger, central colony and not the satellites. The satellite bacteria are unlikely to carry the plasmid that contains the C. elegans lsy-2 gene.
This process of using a marker (usually antibiotic resistance) to differentiate transformed cells from those not transformed is called selection. Because bacteria reproduce asexually and are immobile on solid media, it is likely that the hundreds of thousands of bacteria making up that colony are genetically identical daughters of a single cell. This allows us to take bacteria from a single colony and sub-culture them in liquid media to make millions of identical copies. However, knowing that the bacteria growing in your broth or on your agar with ampicillin all have the plasmid responsible for amp resistance does not confirm that these bacteria also have our gene of interest insert. There are a small proportion of bacteria on your selection plates that may have a plasmid lacking the gene of interest. That can happen when a plasmid is "empty". Our plasmid, pPD129.36 lsy-2, was genetically modified from a purchased base plasmid L4440 genetically synthesized (reference info about pL4440 at | http://www.addgene.org/1654/ . Our vector plasmid pPD129.36 was provided by Dr. Oliver Hobert's lab at Columbia University NY, NY.
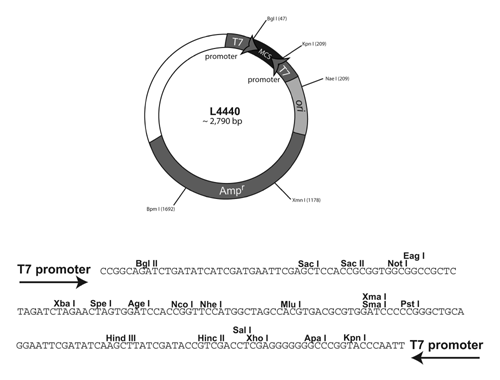
Remember that researchers need to be able to study any gene of interest; therefore, base plasmids are created that allow insertion of any gene of interest in proper alignment with synthetic gene promoters so that the gene of interest can be expressed as desired in a bacterial or a yeast model system. The first step in this process after acquiring a base plasmid (such as L4440) is to isolate small segments of chromosomal DNA that contain your gene. Then you must make lots of copies of your gene by PCR after designing primers that will copy it. The next step is to ligate (insert) the gene from the PCR product into the vector plasmid so that it is in proper alignment with the promoters. If all goes well the ligation works and you achieve this proper alignment and after transformation (uptake of the plasmid by a cell). both the selection gene and the gene of interest are expressed. However, to do a ligation you must cut the vector plasmid at specific places in the DNA using specific restriction enzymes and then reanneal the ends after the gene of interest is inserted. Occasionally that ligation/annealing process fails to work properly and the plasmid DNA anneals back on itself. Since all of that previous work to make pPD129.36 lsy-2 was done by previous researchers, you will begin our project by making sure that you are starting with a bacterial colony that successfully ligated C. elegans lsy-2 gene into the plasmid.
Colony PCR
To confirm that the E coli bacterial colony you picked has lsy-2, we are going to do a colony PCR. Instead of adding purified lsy-2 gene fragments as template DNA in a PCR reaction with primers specific for your gene (as it was done to make the plasmid), you will add a TINY little part of a colony as the template for your PCR reaction. During the first heat cycle the cells will burst open and release their DNA into the reaction. We will test both well-isolated, non-satellite colonies per group to be sure we continue with bacteria that have the gene of interest inserted in our plasmid.
- Obtain a previously prepared culture of E. coli bacteria that have been transformed with pPD129.36 lsy-2 growing on solid Luria broth medium with ampicillin. Select two large, well isolated colonies and label them A and B on the bottom of the plastic plate using your fine tipped Sharpie. Label the plate on the bottom with your initials, your lab day, and add a small strip of your team color tape.
- Obtain three PCR tubes and lids from your instructor in your team color.
- Label the side of each PCR tube A, B and C - the marker WILL rub off the top. Tube C will serve as your negative control with no colony added.
- Add 30 ul of master mix to each tube. Your master mix will include: 23 μL H2O; 3 μL of PCR buffer (10 mM Tris, 50 mM KCl, 1.5 mM MgCl2 pH 8.3); 0.67 μL of 10 mM dNTPs; 0.67 μL of forward primer (20 μM stock); 1 μL of 2 units/μL Taq polymerase. The primer we are using is for T7 polymerase and we are only using one primer rather than two for this amplification. Why do we not need to use both a forward and reverse primer to detect the lys-2 gene in this reaction?
Primer Sequence: specific to T7 RNA polymerase promoter on either side of the lsy-2 gene -
5' TAATACGACTCACTATAGGG 3'
- After each tube has master mix, use the sterile end of an autoclaved toothpick (not the end you are touching) or the end of a sterile micropipet tip and gently touch the center of your colony of interest and pick up a tiny, barely visible amount of the bacteria. DO NOT TAKE THE ENTIRE COLONY!!! Make sure that you select colonies that are large enough to have a significant amount remaining bacteria after taking your sample.
- Gently twirl the toothpick in the tube or mix the bacteria from the pipet tip with the master mix making SURE that you have gotten it off the tip or toothpick and into the reaction. Discard the toothpick or the tip into your orange autoclave bag.
- Repeat for colony B
- Snap the lid on the tubes, pulse them briefly in the microcentrifuge with the appropriate rotor and adapter and bring them to the thermal cycler for PCR initiation.
- Finally, seal the plate with parafilm. Invert it (agar side up so condensation will not drip on your colonies) and store it in the refrigerator in your lab day's rack until the day before the next lab when you will set up an overnight culture from a colony that is positive for lsy-2.
PCR Conditions:
| Step | Temperature | Time | Repeat |
|---|---|---|---|
| 1 | 94°C | 2 minutes | 1 time |
| 2 | 94°C | 30 seconds | |
| 3 | 54°C | 30 seconds | |
| 4 | 72°C | 2 minutes | Steps 2-4 30 times total |
| 5 | 72°C | 10 minutes | 1 time |
| 6 | 4°C | forever | end program |
Agarose Gel Electrophoresis
After the PCR reactions have, we hope, made millions of copies of lsy-2 you will take a sample of the PCR product and run a gel to analyze the results of the amplification (the search for your gene).
- Add 5 μL of loading dye to each PCR product. You will find a precast 1% agarose gel with Sybr-Safe in 1x TAE buffer (prepared by our lab specialist) that is just for two groups' use. Make sure that the wells of the gel are closest to the black (negative) electrode and that the gel apparatus has plenty of buffer. Draw a template in your lab notebook so you know which colony is to be put in each lane (1-7 left to right) and which lane contains your DNA ladder. Make sure you get a copy of the DNA ladder key.
- Load 15μL of each PCR product into separate wells in the gel. Make sure they are loaded in alphabetical order left to right, A, B, C.
- Load 5 μL of the 100 base pair DNA ladder in the well on the far right.
- Run the gel for ~ 20min. at ~120 volts.
Our standards ladder is product number NEB323-2S from New England Biologicals. The manufacturer supplies the following information about the standards ladder.

Capturing Digital Images of Nucleic Acid Gels Stained with SYBR Safe Using the BioRad Imaging System in L308
Instructions for Taking a DNA gel image stained with Sybr Safe using the BioRad ChemiDoc MP System with Image Lab Software
IMPORTANT: Ethidium Bromide stained gels may NOT be imaged in this equipment. Remove gloves and wash hands BEFORE using the computer. DO NOT contaminate the computer. The XcitaBLue Conversion Screen should be kept covering the UV transilluminator and only removed TEMPORARILY when using fluorphores other than Sybr Safe or SybrGreen. Please return the Xcita Blue Screen to the UV transillumintor if you remove it.
Quick and Easy Protocol for photographing Sybr Safe DNA gels:
1) Make sure the Power Button on the right front of the imager shows a green light. If not press it until the green light comes on and wait 5-10 min for warm up.
2) Open the UV transilluminator drawer on the lower front of the imager and make sure that the XcitaBlue Conversion Screen is in place. If not find it and position it covering the glass. Clean it with water and a paper towel if necessary.
3) Position your gel in the center of the glass.
4) Close the drawer, remove gloves, and wash your hands before using the computer to the left of the imager.
5) Open the ImageLab 4.0.1 software by double clicking on the icon on the computer desktop
6) Find and open the Protocol called SybrSafe under Recent Protocols or from the Open Menu. Double click to open it.
7) Click Position Gel (yellow button) to check the position of your gel.
8) Click Run Protocol (green button) to take the photo.
9) To Save your image (if you are not going to analyze it quantitatively), find or make a folder for your work in Documents (NOT on the Desktop). go to File---Export---Export for Publication (use the defaults, e.g. 300dpi). You will see a pop up message reminding you that this option is not optimized for analysis---it doesn’t matter—Click OK. Check the Location where your image will be saved, the FILE Name, and use the drop down menu to SAVE AS TYPE tiff or jpg. Click Save.
10) Close the Image Lab Software 11) Put on gloves, remove your gel, clean the glass with water and a papertowel. 12) The computer AND the ChemiDoc Imager should remain ON. DO NOT Turn OFF the power or shut down or log off the computer.
To do on the day before the next lab:
You and your partner will return to the lab to make an overnight broth culture of one of the colonies that you are sure contains the gene of interest (determined from your visualization of successfully amplified, appropriately sized DNA seen on your gel photo). The sub-culture you will set up tonight will create many identical copies of bacteria that carry the plasmid containing your gene of interest.
- Find your LB+amp plate in the glass front refrigerator in a rack labeled with your lab day. Make sure there is some bacteria remaining on the plate of a colony that you saw successful gene amplification in the PCR product.
- Begin by obtaining two tubes of LB broth (each will have 5 ml of broth) from the refrigerator in the back left hand corner of the room.
- Add 5 microliters of the 50mg/ml ampicillin stock (also found in the refrigerator) to each tube. Calculate the effective concentration of ampicillin that you will have in your LB tube (remember V1 x C1= V2 x C2) and record that information in your lab notebook.
- Add 5 microliters of the 12.5mg/ml tetracycline stock(also found in the refrigerator) to each tube. Calculate the effective concentration of tetracycline that you will have in your LB tube (remember V1 x C1= V2 x C2) and record that information in your lab notebook.
- Gently swirl your LB +amp + tet broth to mix the contents.
- Label the two sterile glass culture tubes with tape in your team color. Label one with "pPD129.36 lsy-2" and your initials. Label the other with your initials only.
- Inoculate the broth with your bacteria by using a sterile disposable loop to scrape your candidate colony off the plate. Be sure not to touch the plate with the loop except on the desired colony and don’t pick up any satellite colonies! Gently swirl the loop in the LB+amp+tet broth - you should be able to see the colony come off the loop. (The second tube of broth labeled with just your initials is a control and should not be inoculated with bacteria as it is your control for contamination.)
- Balance the 2 tubes across from each other on the rotating wheel in the incubator at the front of the room when you come in the door.
- Incubate these broth cultures at 37°C overnight. Do not forget to make sure the wheel is rotating when you leave!
