BIO254:Exam2002

Bio154 Sample Exam 2002-Key
NOTE: Answers are provided by your classmates. Some questions may have multiple correct answers; as long as yours is reasonable, logic and consistent, you can get full credit. Some questions may be challenging but don’t panic; grading will be on a curve. Good luck.
I. (12 pts)
Describe three major similarities and three major differences (biology and/or mechanism) of Rac (a Rho family small GTPase) and Transducin (a trimeric G protein) (The fact that they are both GTPases is a given now.)
Differences:
Rac is a small GTPase and is monomeric Whereas Transducin is heterotrimeric
Rac (small G proteins) can act as a Biological timer. This is because they can convert from the GTP bound form to the GDP bound form almost cyclically. Transducin (heterotrimeric G-proteins) on the other hand, require GDP dissociation factors to dissociate and go back to the GTP bound form. These proteins are called GEFs guanisine nucleotide exchange factors.
Rac plays a role in actin polymerization at the growth cone during development or reorganization. It responds to chemoattractants present in this region. Transducin plays a role in vision consitently throughout life via the conformational change in rhodopsin (mediated by photon absorption) Transducin is membrane associated and works with GPRCs to function in vision
Similarities:
They are both involved in cell signaling and signal transduction They both have alpha, beta and gamma subunits They both get activated by the release of GDP and binding to GTP. They both get deactivated by the hydrolysis or release of GTP and binding of GDP
II. (24 pts)
(a) While searching the human genome you come across a putative receptor for axon guidance based on its conserved motifs with other known axon guidance receptors. You are happy to find out that this receptor has unstudied homologues in other organisms including worm, fly, zebra fish, and mouse. Give two reasons why you should be happy about that (4 pts).
1. The existence of homologues in so many model organisms makes available for experimentation, all the different advantages of each system and the genetic tools that come with them. You have a whole range of possibilities in studying the receptor in different models and comparing their functions.
2. The genetic conservation across species also means that this is most likely a very important gene, that probably also exists in homologous form in humans
(b) You decide to make a loss-of-function mutant of this putative receptor in fly. How will you determine where to look in your mutant animal for phenotypes? (2 pts) Give a reason why you might not see a phenotype even if you have “knocked out” the gene activity completely (2 pts).
i) You could use in situ hybridization to localize the DNA or RNA sequence of your gene.
ii) You may not see a phenotype if the gene you knocked out is redundant. In other words, another gene that is similar enough in sequence or function that it can substitute for your knocked out gene may exist.
(c) Wow! Your mutant has a striking phenotype. You notice that in a subpopulation of neurons your mutant axons fail to make a critical turn at a specific point (neuron A). You are not sure if your newly discovered receptor acts as a receptor for an attractive or repulsive cue. Draw two models postulating an attractive or repulsive cue. (6 pts)

Wild-type Mutant
(d) You also notice that in wild-type animals another subpopulation of neurons never make this turn (neuron B). Propose an experiment to show that your receptor is sufficient for axon turning in this system. (6 pts)

Wild-type Wild-type
(e) You are now interested in what is downstream of your receptor. Propose two general experimental techniques that may help you answer this question. (4 pts)
III. (24 pts)
An interesting mutant of gene X has been found in the course of photoreceptor study. The figure below shows the population mean single photon responses of rods from wild type and from mutant X.

1) According to the picture above, what step of the photoreceptor response is affected by X mutant? (4 pts)
The increased amplitude and prolonged response in the mutant suggests that there is a defect in the deactivation step of the phototransduction cascade. Had the defect been in the activation step, the mutant's activation slope would not align with the wildtype's.
2) What protein could gene X encode? Give three candidates discussed in lectures. Explain how mutations in these three candidate genes could result in the phenotypes seen in the graph. (12 pts)
Possibility #1: Arrestin mutation: Arrestin is necessary to bind phosphorylated rhodopsin in order to fully deactivate it. If arrestin does not work optimally, rhodopsin will take longer to be completely deactivated.
Possibility #2: Heterozygous Rhodopsin Kinase mutant: Activated Rhodopsin on being phosphorylated becomes partially active. The phosphorylation is done by rhodopsin kinase. Only once it is phosphorylated can it be capped by Arrestin and deactivated. In the event that there is not enough rhodopsin kinase produced, it will take longer for the deactivation of rhodopsin to occur giving a response as shown.
Possibility #3: Point mutations in Carboxy terminal tail of rhodopsin molecule: It is required that there be several serine and threonine residues in the carboxy terminal end of the rhodopsin molecule. These are essential for rapid shut off. In the event that there are point mutations and hence a decreased number of serine and threonines, the rate of deactivation would be slower.
Note: proteins that occur too downstream in the deactivation process would not produce the mutant response curve seen in the figure. Because the mutation affects the immediate deactivation steps (deactivation occurs later than it should), downstream proteins such as RGS9 (which is essential to hydrolyze GTP bound transducin to GDP form. GDP bound form is essential to switch off Phosophodiesterase activity and restore dark-state), are not ideal candidates for the mutation. RGS9
3) Come up with two further experiments to distinguish your 3 hypotheses in (2). (8 pts)\
Experiment #1: Create a Rhodopsin kinase KO and a heterozygous animal. Compare the rates of deactivation of the photoresponse amongst these two with the WT. Expect to see a slower deactivation in the heterozygous mutant and no deactivation in the Rhodopsin kinase KO.
Experiment #2: Try to rescue the phenotype. Begin with the most downstream candidate (in this case, arrestin) and try overexpressing it in the mutant. If the rate of deactivation does not increase, eliminate arrestin as a candidate and overexpress rhodopsin kinase. If that doesn't work, the mutation probably exists in rhodopsin itself.
IV. (24 pts)
To investigate the mechanisms of retinocollicular (mammalian equivalent of retinotectal) projection specificity, O’Leary and colleagues generated knock-out mice for both Ephrin B2 and B3 receptors (EphB2 & EphB3). To study the phenotype, they inject an axon tracer DiI into a very small and identical region of the retina in both WT and double knockout mice, and then analyze where these RGC axons terminate in the superior colliculus by following DiI labeled axons. The figure shows the DiI labeled RGC axon termination in the superior colliculus for WT and double knockout mice (L: lateral; M: medial; P: posterior; Anterior border is indicated by the arrowheads).

...............WT....................EphB2-/-;EphB3-/-.....
1) Judging from the location of the termination in the superior colliculus in WT, which part of the retina do you think they inject their DiI? (4 pts)
They inject DiI into the ventral portion of the retina of the eye on the opposite side.
2) In class, we mentioned that EphBs are expressed in RGC in a high ventral-low dorsal gradient; and Ephrin Bs are expressed in superior colliculus in a high medial-low lateral gradient. How can you explain the double knock-out phenotype? (4 pts)
In the absence of EphB as seen in the case of the double knock outs, there is a lateral shift. This is because in the absence of EphB there is no attraction of the growing RGC from the ventral region of the retina towards the Epherin B which is rich in the medial region of the superior colliculus.
3) If EphB2 and EphB3 are the only receptors for D-V patterning, you would imagine that without these receptors, axons will randomly distribute along the D-V axis. However you quickly notice that the ectopic terminal zones (eTZ) always project lateral to the correct terminal zone (TZ) instead diffusely in both directions (see Figure). Propose a simple model that explains this effect. (4 pts)
They are extending branches towards the low level of Epherin B gradient since they do not have EphB. OR The Wnt-Ryk system which works with the EphrinB-EphB system is causing repulsion leading to a eTZ.
4) You may also have noticed that in EphB mutants axons in eTZ largely concentrate on one spot instead of being diffusive. Propose an explanation for this phenomenon and describe a simple experiment to test your hypothesis. (6 pts).
5) It turns out that Ephrin Bs are also expressed in RGC axons in a gradient fashion along D-V axis, and EphB are also expressed in superior colliculus (target) along a M-L axis! Moreover, since the “ligands” for EphBs, the Ephrin B, are transmembrane proteins so they can sometimes act as receptors for “reverse signaling”. So the phenotypes seen in the Figure is not necessarily caused by loss of EphBs in the axons; the loss of EphB in the target could also contribute. Describe one experiment that could distinguish whether EphB is needed in RGC axon or in the target for the correct establishment of topographic map along the D-V axis. (6 pts)
First off, what's reverse signaling? Normally, ligands are free-floating (e.g. hormones), and they bind to a receptor, which initiates some sort of signal cascade. In reverse signaling, the ligand (here, Ephrin B) is a transmembrane protein that remains in the membrane while interacting w/ its receptor (EphB). This means Ephrin B could have its own signal cascade about its interaction with EphB.
The experiment could create mice that lack a cytoplasmic domain of EphB in the RGC axon (meaning the receptor can still interact with Ephrin B outside the plasma membrane, but without the cytoplasmic domain, there would be no transduction cascade for the receptor). If there is an effect, you know EphB is necessary in the RGC axon. If there is no effect, EphB is probably necessary in the target.
You do not want to knockout EphB entirely because if Ephrin B is reverse signaling, effects you may see after the KO could result from the absence of EphB OR the absence of reverse signaling from Ephrin B. By only truncating EphB such that it only lacks its cytoplasmic domain, the remaining part can still interact with Ephrin B and allow for any reverse signaling that may occur.
V. (16 pts)
1) What is the difference between voltage-dependent closure of a channel and inactivation of a channel? (3 pts)
The voltage-dependent closure of a channel occurs at an activation gate when there is a change in voltage. Channel inactivation occurs at the inactivation gate when the voltage is maintained, for example, when the membrane remains depolarized.
2) Describe one example in which inactivation independent of voltage-dependent closure is important. (3 pts)
Inactivation of Na channels is important to limit the duration of an action potential. Upon depolarization, Na channels rapidly open, creating the rising phase of the action potential, but soon after the inactivation gates will gradually close limiting further Na influx.
3) Describe one mechanism by which inactivation is achieved. (2 pts)
In the Na channel, inactivation occurs though a ball and chain model, in which the "ball" blocks the opening of the channel when the inactivation gate is closed.
4) Type-A potassium channels are a group of voltage-gated K channels that rapidly inactivate at membrane potentials at which the classic Hodgkin-Huxley voltage dependent potassium channel stays active.
In the voltage-clamp experiment shown below, a molluscan neuron containing classic Hodgkin-Huxley K channels and type-A potassium channels was depolarized to 5 mV from a holding potential of either -40 mV or -80 mV. The resulting current is quite different: one (from -40mV) consists of only IK (the classic Hodgkin-Huxley channel), the other (from –80mV) consists of IK and IA (the current from the type A channel). One could thus deduce the IA current from subtraction (dotted line).
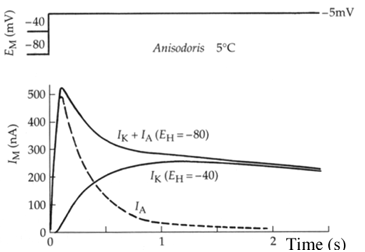

(a) What does this experiment reveal about the properties of the type-A channel? (4 pts)
Given that both channels are open at the spike, it appears that the type-A channel opens first because its line begins practically at t=0, while the classic Hodgkin-Huxley channel opens a few milliseconds later.
(b) Type-A potassium channels prevent neurons from firing repetitively at high frequencies but enable them to fire repetitively at low frequencies. How do the properties of the type-A channel enable it to regulate the spacing of action potentials? (4 pts)
Type-A potassium channels prevent neurons from firing repetitively at high frequencies because firing at high frequencies would raise the cell potential above the voltage that these channels are activated (i.e. -40 mV). At low frequency however, the cell has enough time to reset its voltage down to, say -80 mV), allowing the Type-A channels to continue working.
Recent updates to the site:
List of abbreviations:
- N
- This edit created a new page (also see list of new pages)
- m
- This is a minor edit
- b
- This edit was performed by a bot
- (±123)
- The page size changed by this number of bytes
11 June 2026
| 02:22 | Labs diffhist +52 Mohammad H. Dezfulian talk contribs (→North America) | ||||
|
|
02:14 | User:Mohammad H. Dezfulian 7 changes history −4,949 [Mohammad H. Dezfulian (7×)] | |||
|
|
02:14 (cur | prev) +416 Mohammad H. Dezfulian talk contribs (→Contact Info) | ||||
|
|
02:12 (cur | prev) −269 Mohammad H. Dezfulian talk contribs (→Fellowships) | ||||
|
|
02:12 (cur | prev) −5,151 Mohammad H. Dezfulian talk contribs (→Conference Presentations & Proceedings) | ||||
|
|
02:12 (cur | prev) −880 Mohammad H. Dezfulian talk contribs (→Academic Honors) | ||||
|
|
02:11 (cur | prev) −409 Mohammad H. Dezfulian talk contribs (→Funding) | ||||
|
|
02:11 (cur | prev) +1,357 Mohammad H. Dezfulian talk contribs (→Peer Reviewed) | ||||
|
|
02:07 (cur | prev) −13 Mohammad H. Dezfulian talk contribs (→Contact Info) | ||||
