Angela A. Garibaldi Week 8
Retrieving Protein Sequences
- Go to UniProt UniProt
- Enter dUTPase in search window. This produces more than 3 relevant sequences, so found DUT ECOLI (P06968) on page 4
- Scroll down for FASTA format of amino acid sequences
- In the case that your beginning information is not enough to find the protein sequence you seek,
- find the advanced search option. This no longer exists. You have to click the add and search button and a drop down menu will be displayed to give you the same search options as described in Figure 2-16 of the Bioinformatics for Dummeies

Retrieving a List of Related Protein Sequences
- Go to the Advanced Search UniProt as described above
- Because the advanced search is completely different, cannot deselect TrEMBL. Instead Select Reviewed- Yes as an alternative
- Input dUTPase in search again. There is no "description" field any longer.Yields many possibilities
- Since there are more than 211 total possibilities, so we selected entire first page of sequences (25)
- In newer version click retrieve at the bottom right corner instead of french button.
- Once you retrieve these, it is put into a list of which you can add to and then below choose the format you want the sequences in. No longer have to copy and paste into a document. FASTA format is available.


Reading a Swiss-Prot Entry
This time we skipped the example and did the activity using HIV gp120.
- Select the Reviewed - Yes. Our overall query to achieve these results: HIV gp120 AND reviewed:yes
- We selected the first option in the list
Entry Name: ENV_HV1H2 Accession Number: P04578
- Scroll down to Sequence Annotation - Region to Look at V3 sequence specifically.
ORFing your DNA Sequences
- Go to NCBI ORF Finder
- Input a DNA sequence for practice
I input the following sequence: >S7V1-1 GAGATAGTAATTAGATCTGCCAATTTCACGGACAATACTAAGACCATAATAGTACAGCTGAATGTATCTG TAGAAATTAATTGTACGAGACCCAACAACAATACAAGAAAAAGTATACCTATAGGACCAGGGAGAGCATT TTATGCTACAGGAGAAATAATAGGGAATATAAGACAAGCACATTGTAACATTAGTAGAGCAAAATGGAAT AACACTTTAAAACAGATAGCTACAAAATTAAGAAAACAATTTGAGAATAAAACAATAGTCTTTAATCAAT CCTCA
Compare your results with the SWISS-PROT entry you found for the protein above to decipher what the output means. ExPASy also has a translation tool you can use here
- Based on the ExPASy tool, the following amino acid sequence was the only viable ORF. All others had stop codons within the first few codons
E I V I R S A N F T D N T K T I I V Q L N V S V E I N C T R P N N N T R K S I P I G P G R A F Y A T G E I I G N I R Q A H C N I S R A K W N N T L K Q I A T K L R K Q F E N K T I V F N Q S S
Working with a single protein sequence
Utilizing Bioinformatics for Dummies pages 159-195
- Go to Expasy
- Click protParam near top of page
- Enter sequence into space provided or by pasting the accession number. DO NOT INCLUDE THE FASTA FORMAT FIRST LINE, ONLY RAW DATA.
- Compute parameters

I saved this file on my personal computer since WetWare does not allow html files. This will give information about the protein, composition, ph, stability, etc
- For a tool to simulate cutting of your protein, use: [1]
Looking for transmembrane segments
- go to Protscale
- Enter your sequence in raw format or swiss-prot accession number.
- Select the radio button.
- Choose 19 in the pull-down menu because this number is best for looking for transmembrane helices. 7-11 would be better for globular proteins.

- strong signals are not sensitive to parameters. Recommended threshold for Kyte and Doolittle is 1.6. If you forget this number do the following:
- Place paper over your results.
- Lower the paper until the tips of the strongest peaks appear
- Keep lowering this threshold as long as you can see nice sharp peaks.
- 6 of the 7 transmembrane regions are easy to find.
- Go to TMHMM only FASTA format is recognized
- Keep Output Format radio buttons to their default value.

- This predicts segments that are inside the cell AND segments that are outside of the cell. but fails to predict the segment in the middle, but gives good estimation for 5 of the segments.
Coiled Coil regions
- go to COILS
Predicting post translational Modifications
- go to PROSITE and compare your protein to other collection of patterns in PROSITE.
- Paste your sequence or acession number into the left box (Proteins to be scanned)
- Uncheck Exclude Motifs with High Probability of Occurrence box.
- Check the Do not scan profiles box
- Scan
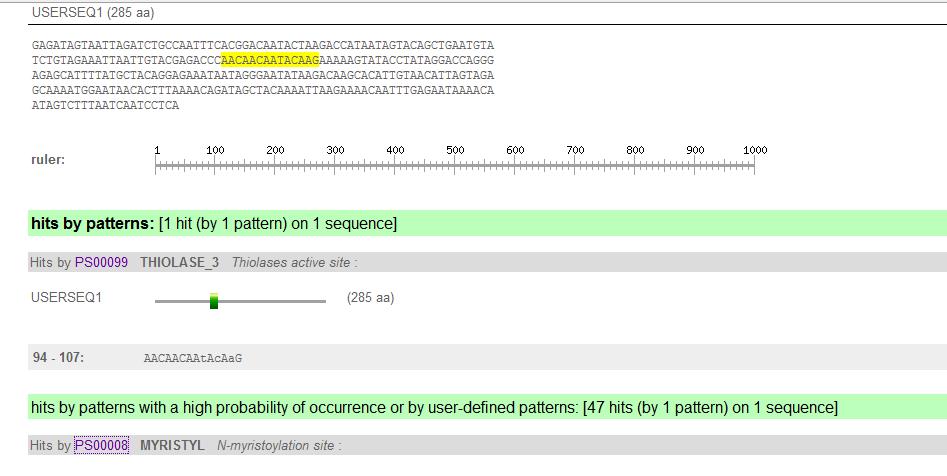
Results:
- Each pattern family has its own color code, with its accompanying details
- PS##### leads to the pattern documentation and information about its biological function
- PDB= Protein Data Base which contains all the 3d structures.
- Click on one of these links for a static GIF picture
- The list has segments containing patterns within your protein. The numbers indicate the position of the match within your sequence, capital letters=residues specified by the pattern; lowercase letters = residues that weren't specified by the pattern
- NOTE: not everything is in PROSITE! Visit expasy tools!

Finding domains with InterProScan
- Go to InterProScan
- Enter your sequence in the search box. Accession numbers do not work
- Choose domain databases you are interested in. TMHMM and SignalPHMM are options that determine transmembrane predictions and single peptide (short N-Terminus segment that causes your protein to reach it's destination in the cell) predictions, respectively.
- Submit job
- Save results via the "Raw Output" button
 Results:
Results:
- First entry in each column indicates the type of diagnosis provided: FAMILY OR DOMAIN
- IPR#### points to the InterPro documentation
- PS#### link will take you to that entry where you can find individual PROSITE documentation
- Colored boxes show you where the match occurred on your sequence
Strengths: Searches multiple databases Weaknesses: domain databases do not agree exactly on the boundaries of the matches.
- See: A common mistake when scanning domains on PAGE 187
Finding domains with the CD Server
- Go to CD server or BLAST and click Search the Conserved Domain Database using RPS-BLAST.
- Paste sequence or its identifier in the box
- Deselect the Apply Low Complexity Filter in the case that there me an over-represented amino acid, making a sequence simpler and losing domains with this simplicity.
- Set the Expect Value Threshold to 1
- Submit ( For this I used a different sequence provided on the CD server site for simplicity)

Results:
- Graphic shows the regions of your protein that match the domain.
- Red domains are from SMART
- Ragged ends indicate partial matches
- E-Value=how many times you can expect this good of a hit by sheer chance. The lower the E-Value, the better. (below 0.01)
- The hit list shows the domains that match your sequence, sorted by E-value. Links lead to documentation.
Finding Domains with Motif Scan
3d structure
- use Cn3dn Cn3D software site
Navigation
Journal Links
Personal Journal
- Angela A. Garibaldi Week 2
- Angela A. Garibaldi Week 3
- Angela A. Garibaldi Week 4
- Angela A. Garibaldi Week 5
- Angela A. Garibaldi Week 6
- Angela A. Garibaldi Week 7
- Angela A. Garibaldi Week 8
- Angela A. Garibaldi Week 9
- Angela A. Garibaldi Week 10
- Angela A. Garibaldi Week 11
- Angela A. Garibaldi Week 12
- Angela A. Garibaldi Week 13
- Angela A. Garibaldi Week 14
- Angela A. Garibaldi GS Papers 1
