20.109(S15):Introduction to cell strains and plating (Day1)

Introduction
Please begin by reading the Module 2 overview.
You will encounter many new concepts and new methods during this module, while building on some of the things you have learned already. The detailed component descriptions will be rolled out over several days, but let's begin with a high-level overview of the entire module.
As depicted below, in Module 2 you will be concerned with measuring DNA repair, specifically through the non-homologous end-joining pathway, or NHEJ – more about that in the coming week. There are two parameters across which you will compare repair: cell type and damage topology. We will work exclusively with Chinese hamster ovary (CHO) cells; wild-type cells are called K1 while cells deficient in the NHEJ protein Ku80 are called xrs6. You will investigate DNA repair in K1 cells with and without an NHEJ inhibitor of your choice, and in xrs6. You learned in Module 1 that double-stranded DNA ends can be "sticky," or more formally "cohesive," (with overhangs) or blunt. In Module 2 we will take things up a notch and investigate about half a dozen different cut topologies: sticky, blunt, sticky on one end and blunt on the other, etc.

The culminating assay we will use for measuring DNA repair is based on paired fluorescent reporters. Briefly, a Blue Fluorescent Protein (BFP) plasmid with a specific type of cut site will be introduced into the CHO cells by similar methods to the bacterial transformation that we performed in Module 1. Simultaneously, an intact Green Fluorescent Protein (GFP) plasmid will be introduced to the cells to measure baseline rates of plasmid uptake during the transfection process. Any cells falling above the bent line (see arrow) are expressing both GFP and BFP, and therefore must have undergone a repair event. By way of background, GFP is naturally produced by jellyfish and was cloned into other organisms in the early 1990’s. It has since been exploited as a genetically encodable reporter and mutagenized to vary its excitation and emission spectra.
To be confident of your findings related to DNA repair, you will validate three components of your system: the deficient cells, the inhibitor function, and the damaged DNA. Briefly, you will: perform a Western protein analysis to assess Ku80 levels in K1 and xrs6 cells; quantify the ability of K1 cells to repair irradiation-induced DNA damage with and without potential inhibitors; verify that the DNA reporter was indeed cut. The validation phase is depicted below.

One major objective for this experimental module is for you to learn how to perform tissue culture. Today you will jump right into this task, and plate CHO cells that next time you will use in a Western protein assay.
Tissue culture was developed about 100 years ago as a method for learning about mammalian biology, and since that time we have learned a tremendous amount by studying the behavior of mammalian cells maintained in the laboratory. The term tissue culture was originally coined because people were doing exactly that, extracting tissue and letting it live in a dish for a short time. Today, most tissue culture experiments are done using cells rather than tissues. Much of what we know about cancer, heritable diseases, and the effects of the environment on human health has been derived from studies of cultured cells.
What types of cells do people study, and where do they come from? Cells that come directly from animal tissue are called primary cells. They are difficult to culture, largely because primary cells in this context divide only a limited number of times. This limitation on the lifespan of cultured primary cells, called the Hayflick limit, is a problem because it requires a researcher to repeatedly remove tissues from animals in order to complete a study. Cell isolation processes can be quite labor-intensive, and also can complicate data analysis due to inherent animal-to-animal variation. To get around this problem, people have studied cells that are immortal, which means that they can divide indefinitely. Some inherent cell-to-cell variation still exists in such populations. Moreover, the genetic changes that cause immortality may affect experimental outcomes. The CHO cell line is an example of a derived immortal population.
The art of tissue culture lies in the ability to create conditions that are similar to what a cell would experience in an animal, namely 37°C and neutral pH. Blood nourishes the cells in an animal, and blood components are used to feed cells in culture. Serum, the cell-free (and clotting-factor free) component of blood, contains many of the factors necessary to support the growth of cells outside the animal. Consequently, serum is frequently added to tissue culture medium, although serum-free media (also called chemically defined media) exist and support some types of cultured cells.
Cultured mammalian cells must grow in a germ-free environment and researchers using tissue culture must be skilled in sterile technique. Germs double very quickly relative to mammalian cells. An average mammalian cell doubles about once per day whereas many common bacteria can double every 20 minutes under optimal conditions. Consequently, if you put 100 mammalian cells and 1 bacteria together in a dish, within 24 hours you would have ~200 unhappy mammalian cells, and about 100 million happy bacteria! Needless to say, you would not find it very useful to continue to study the behavior of your mammalian cells under these conditions!
Protocols
Part 1: Introduction to tissue culture
We'll start with pre-lab lecture: a brief overview of the Module 2 experiment, an introduction to the structure of a paper, and a discussion of concepts and pragmatics related to doing tissue culture.
Afterward, half of the class will begin with Part 2 and half will begin with Part 3 below, since we cannot all fit in the tissue culture room at once.
Part 2 (or 3): Plating cells for next time
We will begin with a brief demo about sterile technique and how to use the tissue culture hoods.
Overview
Each team will be given two T25 culture flasks: one with CHO-K1 cells, and one with xrs6 cells. You will enzymatically detach these adherent cells, count them, and plate them at a specific seeding density in a 6-well culture plate. Next time, after these cells have doubled a couple of times, you will lyse them to make cell free extracts and begin your first assay of the module. For this reason, it is important that you take care to use sterile technique today.
It is essential that you do not mix up or cross-contaminate the K1 and xrs6 cell lines! We suggest that one partner is responsible for each flask, and that partners do not share Pasteur pipets or other equipment.
Protocol
- Each tissue culture hood is partly set up for you. Finish preparing your hood according to the demo, first bringing in any remaining equipment you will need, then picking up the pre-warmed reagents from the water bath, and finally obtaining your cells. Don't forget to spray everything down with 70% ethanol.
- One of the greatest sources for TC contamination is moving materials in and out of the hood since this disturbs the air flow that maintains the sterile environment inside the hood. Anticipate what you will need during your experiment to avoid moving your arms in and out of the hood while your cells are inside.
- Look at your cells as you remove them from the incubator. Look first at the color and clarity of the media. Fresh media is reddish-orange in color and if the media on your cells is yellow or cloudy, it could mean that the cells are overgrown, contaminated, or starved for CO2. Next look at the cells on the inverted microscope. Note their shape and arrangement in the dish and how densely the cells cover the surface.
- After you look at your cells, take the flask to your tissue culture hood to begin the seeding process.
- Aspirate the media from the cells using a sterile Pasteur pipet.
- Wash the cells by adding 3 ml PBS using a 5 mL pipet. Slightly tip the flask back and forth to rinse all the cells then aspirate the PBS with a fresh Pasteur pipet.
- To dislodge the cells from the dish, you will add trypsin, a proteolytic enzyme. With a 2 ml pipet, add 1 mL of trypsin to the flask. Be careful not to pull up the liquid too quickly or it will go all the way up your pipet into the pipet-aid!
- Tip the flask in each direction to distribute the trypsin evenly. Incubate the cells at 37°C for 3-5 minutes, until the cells round up and are easily dislodged from the plate by tapping. Check your cells using the microscope to ensure they are dislodged.
- While you are waiting, you may label the 6-well plate that you will share with your partner. Include your group color and today’s date on the left-hand side (assuming you are right-handed). Then label the two rightmost wells, which you will use for your cells, directly with the cell strain name and the initials of the partner who prepared those particular cells.
- Prepare two 15 mL conical tubes as well.
- Finally, this is a great time to clear out your trash!
- After retrieving your cells, add 3 ml of media to the trypsinized CHO cells and pipet the liquid up and down (“triturate”) to remove the cells from the plastic and suspend them in the liquid.
- Remember: Neither take up nor release quite all the liquid, in order to avoid bubbles.
- When you are satisfied that the cell suspension is homogeneous, transfer a known volume (such as 3.5 mL) to the appropriately labeled 15 mL conical tube. Take 90 μL from this solution into a (labeled!) eppendorf tube. Be sure to keep the conical tube containing your cells capped while you work at the microscope.
- Add 10 μL of Trypan blue to the eppindorf tube with your cells and pipet up and down.
- With a new pipet tip, transfer 10 μL of the cell/dye suspension to a hemocytometer, as shown to you by the teaching faculty. Keep K1 on top and xrs6 on the bottom, so you don't forget which is where.
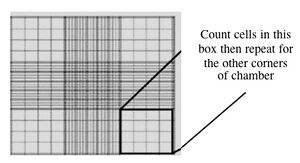
Counting cells using a hemocytometer - Count two diagonal corners, as shown to you by the teaching faculty. If the numbers are within 20% of each other, continue. If they are more different than that, count the other two corners. Be sure to record all of your raw data. Finally, take the average of either the two or the four values.
- This slide has an etched grid of nine large squares. The concentration of cells in a sample can be determined by counting the cells that fall within one such square and then multiplying by 10,000 to determine the number of cells/mL in the solution measured. (Always remember to account for the dilution to cell stock at the end!)
- Note that different squares are sub-divided into different grids. Very dense cells could be counted in the fine grids. In your case, the 4x4 grids and a 10x magnification will be most convenient for counting.
- Calculate the volume of cells you need to seed 50,000 cells in the dedicated well of the 6-well plate.
- For example, if your concentration is 1 million (1 M) cells/mL, you would take 0.05 mL of cells and 2.95 mL of media.
- Make this new cell suspension in a 15 mL conical tube -- first add your cells and then add the needed media. Pipette up and down to mix, but don't introduce bubbles.
- Please note; you may need to mix your original cell suspension by pipetting before distributing the small volume of cells to the new 15 mL conical. Pay attention to see if the cells have settled to the bottom of the conical while you were counting.
- Add the full 3 mL of cell suspension to the dedicated well in your 6-well plate.
- Finally, use the squeaky technique taught in pre-lab to distribute the cells evenly in the plate. Remember to move vertically and horizontally rather than in a circular fashion.
- Waste disposal, final: Aspirate any remaining cell suspensions to destroy them and then clean up the hood. Dispose any vessels that held cells in the burn box, and any sharps in the mayo jars. The next group who uses your hood should find the surfaces wiped down and free of equipment that you brought inside. Please do leave the equipment that was already there.

Part 3 (or 2): Getting familiar with Module 2 cell lines
Before or after you plate the two CHO cell lines today, let's learn a little about their characteristics.
The wild-type strain is called K1, and a great place to learn about this common cell line is through the American Type Culture Collection, or ATCC. Visit the K1 homepage at this link and answer the questions below in your notebook.
- When were CHO cells originally derived?
- Do these cells float in the culture media, or stick to the culture dish? How do you know?
- From what type of tissue (not what organ; e.g., connective tissue) did these cells come? How do you know?
- Finally, look at the two CHO cell images. Which one looks more like the cells you cultured/will culture today? Are there any differences between what you saw and this image?
The NHEJ-deficient strain is called xrs6, and you will learn about it directly from the primary literature, specifically a 1997 paper from Professor Penny Jeggo's lab (PubMed entry linked here). Note: the .pdf file does not download reliably using Firefox, so Safari is a safer browser to examine the paper.
- Introduction
- Read the first paragraph for context and to learn why we care about DNA repair.
- Read the second paragraph through the words "predicted to be radiosensitive." Draw a picture that includes a DNA double-strand break, Ku70, Ku80, and DNA-PKcs, depicting their relationships to each other.
- Read the third and fourth paragraphs for further context about xrs cell lines and for a preview of the major results in the paper. Don't worry about understanding every detail of the fourth paragraph yet – it will make more sense as you explore the results. For now, address the following:
- What protein is defective in xrs6? What gene encodes for that protein?
- Results
- Look at Figure 2 in the paper, in which protein levels are measured by Western analysis. Similarly to a DNA gel, band intensities correlate with protein amount.
- What do you expect to see when you perform your Ku80 Western using K1 and xrs6 cells next week?
- What do we learn from the final lane in Figure 2A?
- Look at Figure 6 in the paper, and its associated text in the "Sequence analysis… (iii) xrs6" section of the paper.
- What is the nature of the sequence change in xrs6 mutants? (Insertion, deletion, substitution? How big?)
- Normally, Ku80 is more than 700 amino acids long. How long is the defective Ku80, and why?
- Skim the section "Analysis of azacytidine-induced revertants," particularly the first and third paragraphs.
- Briefly, why can xrs6 cells revert to a wild-type phenotype?
- Is the defective Ku80 absent from reverted cells?
- Look at Figure 2 in the paper, in which protein levels are measured by Western analysis. Similarly to a DNA gel, band intensities correlate with protein amount.
Homework
- Your Mod1 Abstract & Data Summary is due on Monday, March 16 at 5pm.
- Your first blog post is due within 24 hours of submitting your Abstract & Data Summary. Look for an email from Shannon inviting you to the blog.
- Please note: you need to accept the invitation to the blog within a few days of receipt or it will expire!
We will NOT collect your final worksheet this time; it is only for your benefit.
- Next time you will perform an assay to measure total protein concentration in your cell lysate. The reagent that we will use absorbs light at 600 nm, and the amount of protein present in the lysate will be calculated as:
- protein concentration (μg/mL) = 100 * A600 * 10
- where 10 = the dilution factor used to prepare the assay
- and 100 = the conversion factor from absorbance A to concentration
- To save time during the next lab, you should prepare an Excel spreadsheet that will automatically calculate the volume of your lysate needed to provide between 10-20 μg of total protein.
- The variable input to the sheet will be your measured A600 value.
- Your sheet will be most versatile and easy to follow if each element (such as the dilution factor) is listed and labeled separately, including units.
- The first output of your sheet will be lysate volume. Consider that you will probably want to convert the volume to μL right away.
- Note: you will collect two samples on M2D2. You want to load the same mass of lysate on your gel for both samples.
- When preparing your SDS-PAGE analysis, you may only load a total volume of 24 μL on the gel and 4 μL of that volume will be sample buffer (similar to the sample buffer you used in your DNA electrophoresis). Therefore, your spreadsheet should also calculate the amount of water required to adjust the total volume to 24 μL.
- Try doing your calculations with an A600 of 0.71. If you'd like to check your answer in advance, just email Shannon (skalford at mit dot edu) and she'll let you know if your spreadsheet works.
- protein concentration (μg/mL) = 100 * A600 * 10
Reagent list
- CHO cells
- K1: plated at 200,000 cells per flask 1 day in advance (T/R) or 100,000 cells per flask 2 days in advance (W/F)
- xrs6: plated at 200,000 cells per flask 1 days in advance (T/R) or 100,000 cells per flask 2 days in advance (W/F)
Media components from Life Technologies, Inc. unless noted otherwise.
- CHO cell media
- Dulbecco's Modified Eagle's Medium (DMEM)
- High glucose
- With L-Glutamine
- 10% fetal bovine serum (FBS) from Atlanta Biologicals
- 100X antibiotic solution from Cellgro
- 10,000 U/mL Penicillin
- 10,000 μg/mL Streptomycin
- 100X non-essential amino acids (NEAA)
- varying amounts of seven amino acids
- Dulbecco's Modified Eagle's Medium (DMEM)
- Dulbecco's phosphate-buffered saline (D-PBS)
- 0.25% Trypsin/0.91 mM EDTA
Navigation Links
Next Day: Begin Western protein analysis
Previous Day: Journal club II
