20.109(S09): siRNA design (Day1)
_frontpg.JPG)
Introduction
In the previous experimental module, your work focused on proteins, and to implement protein modifications, you manipulated DNA. In this experimental module, RNA gets the spotlight. While DNA almost exclusively functions to encode the genetic information of a cell, RNA is remarkably versatile. The scientific literature on RNA includes reports of enzymatically active RNAs called ribozymes (indeed, our DNA/RNA/protein world is thought to have evolved from an RNA-based one), and RNAs that directly regulate transcription and translation. RNA has long been used as a readout for gene expression but has only recently been appreciated as a tool for manipulating gene expression.
The term “gene expression” does not refer to happy faces on the DNA as the name implies but is a term used to describe how much of a gene product is synthesized by a cell. In liver cells, expression of genes for liver-specific proteins is high and that of brain-specific genes is low. Many diseases arise from mis-expression of genes. For example, cancer cells make lots of proteins they shouldn’t and grow without limit because the normal regulators of gene expression are broken or malfunctioning.
Gene expression is often regulated at the level of transcription and examples of transcriptional regulation are numerous. The bacterial lac operon that you encountered in Module 1 is the classic example, using DNA-binding proteins to both enhance and repress transcription of the operon. However, many examples of post-transcriptional regulation exist. Recently, studies in worms (C. elegans), fungi (e.g. N. crassa), plants (A. thaliana) and flies (D. melanogaster) revealed a mechanism of gene silencing called RNA interference (RNAi), in which repression is mediated by double stranded RNA (dsRNA). The powerful genetic tools available for these organisms led to the rapid identification of many genes important for gene silencing by dsRNA. RNAi studies have progressed rapidly through a combination of genetics, molecular biology and biochemistry to become one of the most exciting areas in gene expression research.

RNAi can silence particular genes in mammalian cells although other expression effects, not specific for the targeted gene, can be seen. Mammalian cells seem to interpret dsRNA as a viral infection and initiate a response to protect themselves from it. Shorter dsRNAs, sensibly called short interfering RNAs (“siRNAs”) are 19-25 nucleotides long and can bypass the cell’s surveillance system. Indeed, siRNA has silenced genes in many types of cultured mammalian cells, including neuronal, epithelial and fibroblast cells.
The biochemistry of the RNAi pathway is relatively well characterized and the general features of siRNAs are known. To induce RNAi, a dsRNA is processed within the cell to an siRNA which then binds to its single-stranded mRNA target. This interaction stimulates the destruction of the mRNA, effectively silencing gene expression.
After processing, siRNAs possess a sense and an antisense strand. Their 3’ ends overhang by 2 bases and each strand has a 5’ phosphate group and a 3’ hydroxyl. By convention, the siRNA duplexes are described by the sense strand, with the first position of the 5’end referred to as position 1. This usually corresponds to position 19 or so on the antisense strand.
In this experimental module, RNAi will be used to silence gene expression in a mammalian cell line. Today you will design an siRNA to silence luciferase, a gene not normally found in the cell line we’ll study, and also get some practice with cell culture. Later in this experiment, you will simultaneously introduce the luciferase gene and your siRNA into cells, then observe RNAi in action through a simple luminescence assay. You will also use a commercially validated siRNA against a mouse gene of interest to observe direct, indirect, and potentially off-target effects of an siRNA via microarray.
Protocol
Half the class today will start in the cell culture facility and half will start with siRNA design. Midway through class, you'll switch places.
Part 1: siRNA design
Although the biochemistry of dsRNA processing is well understood, less is known about the features that make some siRNAs potent silencers of gene expression and other siRNAs useless. Many times researchers will design four or more siRNAs for a target gene and find only half of them work well. In designing siRNAs, the messenger RNA’s sequence must be known, but choosing which region to target is mostly guesswork. The siRNA sequence must bind an invariable region of the gene. It has been reported that a one basepair mismatch between the target and siRNA can convert an effective inhibitor into a useless one. Conversely, siRNAs can work promiscuously and silence non-target genes, leading to effects on genes that bear some sequence similarity to the targeted one. Some reports suggest that as little as 14 base pairs of complementarity can cause an siRNA to silence an off-target gene.
Design of siRNA against Renilla luciferase
Renilla reniformis is a soft coral, often called a sea pansy, which washes up on Florida beaches after a storm. It will bioluminesce when disturbed due to GFP and a gene for a second light-making protein, luciferase. The biochemistry of the luminescence will be described in detail later in the module. Today, you and your partner will examine Renilla’s gene for luciferase and will be assigned one portion to target for RNAi.
| Team Color | Target | Team Color | Target |
|---|---|---|---|
| Red | R_luc bp 1-156 | Pink | R_luc bp 781-936 |
| Orange | R_luc bp 157-312 | Purple, T/R | R_luc bp 1-156 |
| Yellow | R_luc bp 313-468 | Grey, T/R | R_luc bp 157-312 |
| Green | R_luc bp 469-624 | Purple, W/F | R_luc bp 469-624 |
| Blue | R_luc bp 625-780 | Grey, W/F | R_luc bp 625-780 |
Begin by retrieving the sequence of the gene you hope to silence. Open a web browser program and go to the Promega homepage. The Renilla luciferase gene was fully sequenced in 1991 but the clone you will study is expressed on a plasmid that is commercially available from Promega.
Search the site for psiCHECK2, the name of the plasmid with the Renilla luciferase gene. The “psicheck2 vector” link will retrieve the plasmid sequence with some landmark information at the top of the page. Copy the Renilla luciferase gene sequence into a new MSWord document (remember that the sequence should begin ATG and end with a stop codon; it will be 935 bases long) then trim the sequence to the area of the gene you have been assigned to target. This direction is in bold because people in the past have forgotten to do this!!
Open a new browser window and go to the Ambion homepage. Like Promega, Ambion is a life sciences company selling many useful products for biological research. Ambion is particularly well regarded for its support of RNAi technology. You will use their search algorithm to assist in your siRNA design. This algorithm is found through their “RNAi resource” link. Under “siRNA design tools” you can click on “siRNA Target Finder” to get started. This page has lots of important information to read and good links to follow.
When you are ready to begin the design of your siRNA, paste your sequence from the MSWord document you started into the box that is near the bottom of the webpage. Delete any numbers once you’ve pasted the sequence. Choose “ends with TT.” Choose “all G/C contents.” Since your siRNA will be chemically synthesized, constrain the sequence to avoid 4 or more Gs or Cs in a row.
After you submit your query, several target sequences will appear as a list at the bottom of the webpage. The candidate sequences are listed according to where they bind the target mRNA, one parameter that has been shown to have no consistent or predictable effect on siRNA efficacy. To decide which siRNA candidate on the list is most likely to silence luciferase specifically, copy each target sequence into a separate cell of a new Excel worksheet. You should also paste the sequence you queried.
For each siRNA candidate sequence, consider and record the following:
- What is the G/C content of the candidate? 30-50% G/C tends to work best. Some of the programs used in the next step will also return information on the G/C content of your sequence.
- Is the sequence likely to form secondary structures? These might make the siRNA difficult to process or unlikely to bind its target mRNA. The easiest predictors of secondary structure are the melting temperature (Tm) of the dsRNA and the free energy of the sequence.
- Tm can be calculated by pasting the sequence into the Oligo Analyzer web-program available through Integrated DNA Technologies. Be sure the target type is set to RNA. You can also perform secondary structure analysis using the Hairpin function.
- There are several other web-based programs available to predict RNA secondary structure and calculate its free energy. One such program is here. Another site to use for comparison is OligoCalc which will return Tm, G/C content and free energy calculations.
- Be sure to observe whether a given site is displaying the free energy of specific folded RNA structures, or of the RNA oligo binding to complementary DNA. You would like the RNA binding to DNA to be highly favourable, but the RNA folding to be unfavourable (relatively speaking).
- What is the similarity of each target sequence to other mRNAs in mouse cells? Such sequence similarity is an important consideration since an siRNA can be specific for its target only if it fails to base pair with mRNAs from other genes. The Ambion list of candidate sequences has a BLAST link for each target sequence. Follow each link and query the "Mouse genomic + transcript" database since these siRNAs will be transfected into a mouse cell line. For each BLAST result scroll through the list for any mRNAs that have 16-17 contiguous bases of homology with the siRNA candidates and enter the identity of these genes into your Excel spreadsheet. Note that the genes are listed from most to least homologous, and that the "Max score" indicates a certain degree of homology - for example, 26.3 indicates about 13 bp of homology - so you do not need to individually click on or scan each gene, just the top few.
You should now be able to identify the best siRNA candidate sequence. Please print out one copy of your Excel spreadsheet, circle your choice of siRNA, and turn it in with your notebook.
Part 2: Introduction to cell culture
Background
In the past century, we have learned a tremendous amount by studying the behavior of mammalian cells maintained in the laboratory. Tissue culture was originally developed about 100 years ago as a method for learning about mammalian biology. The term tissue culture was originally coined because people were doing exactly that, extracting tissue and letting it live in a dish for a short time. Today, most tissue culture experiments are done using cells rather than tissues. Much of what we know about cancer, heritable diseases, and the effects of the environment on human health has been derived from studies of cultured cells.

What types of cells do people study, and where do they come from? Cells that come from a tissue are called primary cells, because they come directly from an animal. It is very difficult to culture primary cells, largely because primary cells that are placed in culture divide only a limited number of times. This limitation in the lifespan of cultured primary cells, called the Hayflick limit, is a problem because it requires a researcher to constantly remove tissues from animals in order to complete a study. Cell isolation processes can be quite labour-intensive, and also can complicate data analysis due to inherent animal-to-animal variation. To get around this problem, people have studied cells that are immortal, which means that they can divide indefinitely. (Some inherent cell-to-cell variation still exists in such cells.)
One type of familiar immortalized cell is the cancer cell. Tumor cells continuously divide allowing cancer to invade tissues and proliferate. Cancer cells behave the same way in culture, and under the right conditions, cells can be taken from a tumor and divide indefinitely in culture. Another type of immortalized cell is the embryonic stem cell. Embryonic stem cells are derived from an early stage embryo, and these cells are completely undifferentiated and pluripotent, which means that under the right conditions, they can become any mammalian cell type. Mouse embryonic stem cells have become a valuable research tool, and it is this cell type that we will be using for our current experimental module.
The art of tissue culture lies in the ability to create conditions that are similar to what a cell would experience in an animal, namely 37°C and neutral pH. Blood nourishes the cells in an animal, and blood components are used to feed cells in culture. Serum, the cell-free (and clotting-factor free) component of blood, contains many of the factors necessary to support the growth of cells outside the animal. Consequently, serum is frequently added to tissue culture medium, although serum-free media exist and support some types of cultured cells.
Cultured mammalian cells must grow in a germ-free environment and researchers using tissue culture must be skilled in sterile technique. Germs double very quickly relative to mammalian cells. An average mammalian cells doubles about once per day whereas many common bacteria can double every 20 minutes under optimal conditions. Consequently, if you put 100 mammalian cells and 1 bacteria together in a dish, within 24 hours you would have ~200 unhappy mammalian cells, and about 100 million happy bacteria! Needless to say, you would not find it very useful to continue to study the behavior of your mammalian cells under these conditions!
One major objective for this experimental module is for you to learn how to perform tissue culture. You will learn how to get mouse embryonic stem cells to grow in a dish and also how to prevent contaminants from getting into your cell cultures.
Protocol
Each of you will have a 35 mm dish of mouse embryonic stem (MES) cells that you will use to seed a six-well dish. You and your partner will seed the dishes at different concentrations so you should decide who will seed at 1:5 and who will seed at 1:2.
- Prepare your tissue culture hood according to the demo, bringing in any equipment you will need, then picking up the pre-warmed reagents from the water bath. Don't forget to spray everything down with 70% ethanol.
- One of the greatest sources for TC contamination is moving materials in and out of the hood since this disturbs the air flow that maintains the sterile environment inside the hood. Anticipate what you will need during your experiment to avoid moving your arms in and out of the hood while your cells are inside.
- Look at your cells as you remove them from the incubator. Look first at the color and clarity of the media. Fresh media is reddish-orange in color and if the media on your cells is yellow or cloudy, it could mean that the cells are overgrown, contaminated or starved for CO2. Next look at the cells on the inverted microscope. Note their shape and arrangement in the dish and how densely the cells cover the surface.
- Aspirate the media from the cells using a sterile Pasteur pipet. Dip the pipet in your beaker of ethanol when needed (to clean it).
- Wash the cells by adding 2 ml PBS using a 5 mL pipet. Slightly tip the dish back and forth to rinse all the cells, and then aspirate the liquid.
- To dislodge the cells from the dish, you will add trypsin, a proteolytic enzyme. Using a 2 ml pipet, add 0.7 ml of trypsin to the flask. Be careful not to pull up liquid too quickly or it will go all the way up your pipet into the pipet-aid!
- For one minute precisely (use your timer), tip the flask in each direction to distribute the liquid evenly, then aspirate all but a thin coating of the trypsin off the cells. Incubate the nearly dry cells at 37° for 10 minutes, again using your timer to precisely time this incubation.
- While you are waiting, you can add 1 ml of gelatin to each well of a six-well dish (one dish per pair). This should be done in the sterile hood with sterile technique.
- The gelatin will be removed before you seed the dish with your MES cells but it is important to pre-treat the dish this way. The gelatin must remain in the wells for at least 10 minutes.
- With a 5 ml pipet, add 1.3 ml of media to the trypsinized MES cells and pipet the liquid up and down (“triturate”) to remove the cells from the plastic and suspend them in the liquid. Remove a small amount of the suspension (perhaps 50 μL) to an eppendorf tube and take it to the inverted microscopes.
- Fill one chamber of a hemocytometer with 10 μL of the cell suspension to count the cells.
- This slide has an etched grid of nine large squares. The square in the center is further etched into 25 squares each with a volume of 0.1 ul and 16 tiny chambers (4x4 pattern). The concentration of cells in a sample can be determined by counting the cells that fall within the 4x4 pattern and then multiplying by 10,000 to determine the number of cells/ml.
- You should count the cells in the four corner squares of the 25 square grid, then average the numbers to determine the concentration of cells in your suspension. Save your raw data for the Day 2 FNT assignment!
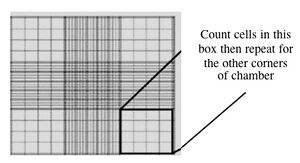
Counting cells using a hemocytometer
- You and your partner will seed at different concentrations. Decide if you will try the 1:5 or 1:2 dilution and add the appropriate amount of cell suspension to 9 ml of media in a 15 ml conical tube.
- Note that the dilution factor refers to the volume of the original cell culture, not the volume that you are moving the cells into. (If the surface area of the 35 mm dish and 6-well dish were very different, we would also want to take that into account.)
- Remove the gelatin from the six-well dish you have prepared and add 3 ml of your cell dilution to each well. Be sure to label your dish with your name, today’s date, the cell line (called “J1”) and the type of media you have used. Return your cells to the incubator.
- Aspirate any remaining cell suspensions to destroy them and clean up the hood. Dispose any vessels that held cells in the Biohazard waste and any sharps in the grey bins. The next group who uses your hood should find the surfaces wiped down, no equipment left inside, the sash closed.

DONE!
For next time
- Read the following articles for a class discussion next time. You will have 45 minutes of in-class time to complete and review your reading. As before, each pair of students will present one figure or so to the rest of the class.
- The news story by Erika Check called "RNA interference: hitting the on switch" published in Nature 2007 vol. 448 pp. 855-8
- The original paper describing some work labeled "RNA activation" by Long-Cheng Li, et al. The article is titled: Small dsRNAs induce transcriptional activation in human cells and was published in PNAS 2006 vol. 103 pp. 17337-42.
- Questions to guide your reading and some background information can be found on the page associated with the next lab. You do NOT have to turn in the answers to these questions but you will be asked to guide the class discussion around some of these topics so you must be familiar with the content of the articles and ready to discuss the material. Again, you will have 45 additional minutes to prepare during class, in recognition of the fact that close-reading of an unfamiliar technical paper may be a multi-hour process.
- Begin familiarizing yourself with the assignments for this module. Next week you will each be presenting a research article to the class. You can sign up for a time and choose an article here. Please send your article choice to me by 24 hrs before your presentation. You will also have a short written assignment in conjunction with the presentation, described here.
Reagents list
- Phosphate-Buffered Saline (PBS)
- 150 mM sodium chloride
- 5.5 mM dibasic sodium phosphate
- 1.0 mM monobasic potassium phosphate
- pH 7.4
- Trypsin solution
- 0.25% Trypsin in HBSS, 1 mM EDTA
- Gelatin solution
- 0.1% TC-grade gelatin prepared in H2O
- Gelatin is a protein prepared by partial hydrolysis of collagen
- J1 ES Cell Culture Medium
- DMEM (high glucose = 4.5 g/L), with supplements listed below
- 2 mM L-Glutamine
- 100 U/ml Penicillin/Streptomycin
- 0.1 mM β-ME
- 0.1 mM Non-Essential Amino Acid (NEAA)
- 15% Fetal Bovine Serum (Atlantic Biologic, Inc., Atlanta, GA)
- Leukemia Inhibitory Factor (LIF), 1000U/mL - LIF helps stem cells maintain their undifferentiated state
- HBSS (Hank's Balanced Salt Solution), calcium/magnesium-free
- 0.4 g/L potassium chloride
- 0.06 g/L monobasic potassium phosphate
- 0.048 g/L dibasic sodium phosphate
- 8.0 g/L sodium chloride
- 0.35 g/L sodium bicarbonate
- 1.0 g/L D-glucose
- 0.01 g/L phenol red
- pH 7.0-7.4
