20.109(F10): Mod 1 Day 8 FACS analysis

FACS analysis
Introduction
Today you will be using flow cytometry to measure the percentage of cells that are fluorescent. You have lipofected cells with two non-functional EGFP genes. Recombination between these two genes can restore the full length EGFP coding sequence so that cells express EGFP. By measuring the percentage of cells that fluoresce green, you will have some measure of the frequency of homologous recombination within mammalian cells. We will use the BioAnalyzer from Agilent Technologies to measure the fluorescence. This instrument will provide data that is more traditionally acquired through flow cytometry at a FACS machine like the ones found at MIT's Cancer Center.
Traditional FACS and Flow Cytometry
FACS stands for "Fluorescence Activated Cell Sorting." The FACS machine has revolutionized biology by allowing researchers to isolate cells based on their spectral qualities. For example, if you have a fluorescently tagged antibody that preferentially binds to a certain cell type, you can isolate a pure sample of this cell type from a complex mixture by using a FACS machine. In addition to purification, the FACS machine can count the number of cells that have a certain spectral quality. If a FACS machine is used just for counting and not for separating subpopulations of cells, then the procedure is called "flow cytometry." FACS is technically challenging and most FACS machines are only run by experts. In contrast, biologists are often trained to perform flow cytometry in order to analyze the proportion of their sample that has particular spectral qualities.


Before there were FACS machines, there were Coulter counters. Coulter counters are automated cell counting machines developed in the 1950s that count cells as they flow in a liquid stream. In an ingenious conceptual leap, Mack Fulwyler combined the technology of ink jet printers with that of Coulter counters to develop the first FACS machine. The ink jet printer head works by vibrating a nozzle so that a spray of discrete droplets is formed. Similarly, in a FACS machine, a liquid suspension of cells is forced at high pressure through a vibrating nozzle to create tiny charged droplets, each containing a single cell. The stream of droplets pass in front of a laser beam, and the scattered light is analyzed by a series of filters and photomultiplier tubes that convert the light signal into electrical impulses. Thus, each cell is "interrogated."
For FACS, the spectral qualities of the cell are analyzed nearly instantaneously and compared to your desired spectral qualities. For example, if you have a mixture of green fluorescent cells and non-fluorescent cells, you can ask the machine to isolate the green cells. If a cell registers as green, an electrical charge deflects the cell to make it fall into a collection chamber.
The BioAnalyzer
This "in lab' instrument works in ways that are similar to the flow cytometer but uses microfluidics, vacuum pumps, and a disposable chip to present the cells one by one to a detector that measures red and blue fluorescence. A very useful animation of the microfluidics is found here. In short,
-
 Six samples can be loaded on a single chip for analysis
Six samples can be loaded on a single chip for analysis -
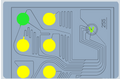 Each sample (shown in yellow) is pumped though a maze of microcapillary tubes to meet with a viscous liquid (shown in green).
Each sample (shown in yellow) is pumped though a maze of microcapillary tubes to meet with a viscous liquid (shown in green). -
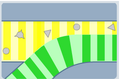 The difference in viscosity between the samples squeezes the cells so they flow one-by-one past an optical detector
The difference in viscosity between the samples squeezes the cells so they flow one-by-one past an optical detector -
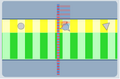 allowing each cell to be queried for "redness" and "blueness."
allowing each cell to be queried for "redness" and "blueness."




Sensibly, the blue detector will measure GFP fluoresence from our cells. To know we are looking at intact, living cells and not fluorescent cell debris, we will also be staining the cells with a special red dye. CellTracker Red can freely diffuse through cellular membranes, and then inside the cytoplasm it is cleaved to an impermeable derivative. Thus living cells with intact membranes retain the dye and fluoresce red, while the disrupted membranes on dead cells do not. By comparing the number of red cells to the number of red and "blue" (i.e. green from GFP) cells in each sample, we'll know the frequency of recombination between the delta5 and the delta3 constructs, as well as the number of green cells in our control samples.
Protocol
Preparing your cells for the BioAnalyzer
The following protocol should be performed in the sterile hoods unless otherwise indicated.
Part 1: Labeling Cells for Viability
The CellTracker Red has been dissolved to 1 mM in DMSO.
- Make sure that the CellTracker solution is completely thawed and then mixed via vortexing.
- Dilute CellTracker Red 1:1000 in DMEM. You will need at least 3 ml for your 6 samples.
- Warm the CellTracker/DMEM solution the 37°C water bath for 5 minutes.
- Wash the cells once with prewarmed 1X PBS.
- Aspirate the PBS.
- Add 0.5 ml of the CellTracker/DMEM to each well.
- Incubate at 37° for 20 minutes
Part 2: Collecting and Counting Cells
This part of the protocol will remind you of what we did on Day 6 of this module.
- Aspirate the CellTracker Red/DMEM from the wells.
- Wash the cells with 1X PBS.
- Add 200 ul of trypsin to each well. Rock in the hood for 1 minute exactly, then aspirate the trypsin
- Incubate the "dry" cells in the incubator for 5 minutes.
- Add 500 ul of DMEM to each well, pipetting up and down to break up any cell clumps
- Move the cells to a 15 ml conical tube.
- Rinse each well with 500 ul of DMEM to recover additional cells and pool with the cells already in the falcon tubes (Pay attention here and don't mix up the cell samples!)
- Remove a 25 ul aliquot of cells to count (dilute 1:2 in DMEM to make # of cells more manageable). You will want 2x10^6 cells/ml for analysis. Look at the Day 6 protocol for this module if you need a refresher for how to do this calculation.
- While counting, spin the falcon tubes 500 g for 5 minutes
- Aspirate the supernatant and resuspend the pellets in the appropriate volume of Agilent's "Cell Buffer" to reach ~2x10^6 cells/ml. Pipet up and down ~10X to mix.
- Keep cells on ice until you are ready to load them onto the BioAnalyzer chip.
Data collection from transfected cells
The BioAnalyzer should be ready for you but you should double check that the chip is in slot position “2” and that the top cartridge is labeled “2." You can choose “cell” and then “flow cytometry” from the “Assay” drop-down menu in the toolbar. You should choose assay = “red to blue” and can use the default settings for time, events to count etc.
To prepare the chip for analysis
- Add 10 ul of "Priming Solution" to the chip in the indicated port
- Wait 1 minute
- Add 15 ul of "Focusing Dye" and 30 ul (X2) "Cell Buffer" to the appropriate ports.
- Add 10 ul of each sample to ports 1-6. Your EGFP transfected sample should be in the 6th (last) port since it is expected to have the greatest number of green cells, and any stray cells from this sample that remain near the detector might throw off your other measurements.
- Load the chip onto the BioAnalyzer and close the cover. Start the run within 5 minutes of loading your samples onto the chip.
- Press the start button in the BioAnalyzer software and all the samples will be analyzed. A full run will take ~25 minutes.
For next time
- Please submit your finished "P3." This assignment is due by 11:00 a.m on 10.12.10. Please turn in your P3 electronically by uploading it to the Stellar website that is associated with our class. It is important that you name your file according to this convention: Firstinitial_Lastname_LabSection_assignment.doc, for example: S_Hockfield_TR_P3.doc There will be a 1/3 letter grade penalty for each day (24 hour period) late. If you are submitting your assignment after the due date, it must be emailed to nlerner, lsutliff, and nkuldell AT mit DOT edu. There will be no re-write option on this assignment.
- If you have not contributed your thoughts, comments and ideas to the 20.109 class blog, remember that you are required to post at least 300 words once each experimental module.
- Begin to familiarize yourself with the content of the second experimental module by reading the front page for the module as well as skimming the introduction to the first day of labwork.
