Biomod/2013/UT-Austin/Methods
<html> <style type="text/css"> // </style>
<a href="http://openwetware.org/wiki/Biomod/2013/UT-Austin"><img src="http://openwetware.org/images/2/2f/G4262.png" width ="50%" height="50%"/>
<a href="http://openwetware.org/wiki/Biomod/2013/UT-Austin/Introduction"><img src="http://openwetware.org/images/0/0d/Intro.png" width="20%" alt="" /></a>
<a href="http://openwetware.org/wiki/Biomod/2013/UT-Austin/Design"><img src="http://openwetware.org/images/0/0a/Design.png" width="10%" alt="" /></a>
<a href="http://openwetware.org/wiki/Biomod/2013/UT-Austin/Methods"><img src="http://openwetware.org/images/0/07/MEthods.png" width="15%" alt="" /></a>
<a href="http://openwetware.org/wiki/Biomod/2013/UT-Austin/Results"><img src="http://openwetware.org/images/1/17/Results.png" width="12%" alt="" /></a>
<a href="http://openwetware.org/wiki/Biomod/2013/UT-Austin/Team Info"><img src="http://openwetware.org/images/3/32/Team.png" width="15%" alt="" /></a>
<a href="http://openwetware.org/wiki/Biomod/2013/UT-Austin/References"><img src="http://openwetware.org/images/0/0d/References.png" width="20%" alt="" /></a>
</html>
Computational Methods
CircDesigNA
CircDesigNA is nucleic acid design software that interfaces with NUPACK to generate sequence for the design of oligonucleotides with a desired secondary structure. The user can specify complementary domains using the nomenclature presented in the previous circuit figures. Additionally, users can specify sequence constraints such as those dictated by our target sequences.
NUPACK
NUPACK is a free web-based software developed by Caltech and is useful for analyzing nucleic acid systems. This program provides information regarding the most thermodynamically stable nucleic acid structures, specifically for DNA on DNA hybridization and RNA on RNA hybridization. There is no information provided for DNA on RNA hybridization.
Geneious
Geneious is a bioinformatics software program. For our purposes it was useful in designing and annotating the MDM2 gene construct.
OligoAnalyzer
The OligoAnalyzer on the IDT website was used to estimate primer melting temperatures for PCR.
Primer Design
Primers were designed using Geneious. Melting temperatures were estimated using IDT’s OligoAnalyzer and unwanted secondary structure was avoided by NUPACK analysis.
Experimental
Oligonucleotide Preparation
All oligonucleotides were ordered from Integrated DNA Technologies (Coralville, IA). Non-HPLC purified oligos were purified through polyacrylamide gel electrophoresis (PAGE) and concentrated by ethanol precipitation.
Page Purification Protocol
Gel Setup
Add 25 mL 8% denaturing acrylamide (containing cold 7 M urea), 25 µL of TEMED, and 100 µL 10% APS into a conical vial. Mix by inverting. Pour the gel solution between the glass plate assembly and insert an electrophoresis comb into the gel. Allow the gel to set before continuing.
Running the Gel
Prepare samples by mixing each DNA sample with an equal volume of 2X denaturing dye. Denature the samples by incubating them at 90°C for 5 minutes. Attach the glass plate assembly to PAGE rig and fill the top and bottom chambers with 1X TBE. Clear lanes of excess urea before loading samples with a transfer pipette. Pre-run the gel at 450 V for 10-20 minutes. Load samples and run the gel at 450 V with a fan.
Visualizing
Remove the gel from the glass and wrap it in plastic wrap. Visualize with UV light and mark the bands of interest.
Excising DNA
Use a new razor to cut out each marked band into a clean conical tube.
DNA Elution following Denaturing PAGE
Add 3 mL of 0.3 M NaOAc to each conical tube. Elute overnight at 37°C in an incubator.
Ethanol Precipitation
Filter the eluted solution using microcentrifuge filter tubes. Aliquot the filtered solution into 400 μL volumes. Add in 1 mL of 100% ethanol and 2 μL of linear polyacrylamide to each aliquot. Mix by inversion before cooling in the -20°C for 30 minutes. Spin down at 13,000 rpm for 10 minutes. Remove supernatant from pellet and wash the pellet with 1 mL of chilled 70% ethanol. Spin down at 13,000 rpm for 5 minutes and then remove the supernatant. Cover and leave out to air-dry. Once dry, resuspend using diH2O.
Target Template Preparation
Apurinic
Apurinic target templates were ordered from IDT and PAGE purified.
BioM_rRNA_Target_Purinic rUrArCrGrCrCrCrCrCrUrCrArGrUrArCrGrArGrArGrGrArArCrCrGrGrGrGrGrUrUrC
BioM_rRNA_Target_Apurinic rUrArCrGrCrCrCrCrCrUrCrArGrUrArCrG/idSp/rGrArGrGrArArCrCrGrGrGrGrGrUrUrC
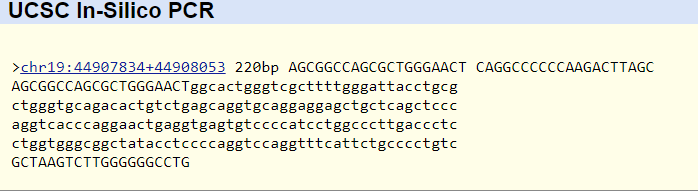
MDM2 Gene Construction
The MDM2 gene excerpt was constructed through overlapping PCR, purified by agarose gel electrophoresis, and concentrated using Promega’s Wizard SV Gel & PCR purification system.
Overlapping PCR
| μL | Conc. | Reagent |
|---|---|---|
| 5 | 10X | AccuPrime Pfx Reaction mix |
| 1 | 20μM | BioM_MDM2_309_Temp1_T_F |
| 1 | 20μM | BioM_MDM2_309_Temp2_F |
| 1 | 20μM | BioM_MDM2_309_Temp3_R |
| 1 | 20μM | BioM_MDM2_309_Rvs |
| 1 | 2.5U/uL | AccuPrime Pfx DNA Polymerase |
| 40 | diH2O | |
| 50 | Total |
| Step # | Temp. (°C) | Time (sec) |
|---|---|---|
| 1 | 95 | 120 |
| 2 | 95 | 15 |
| 3 | 47 | 30 |
| 4 | 68 | 30 |
| 5 | Go to step #2 49 times. |
Agarose Gel Purification Protocol
1. Loosen the lid on a bottle of agarose (2% NuSieve + EtBr) and melt in the microwave.
2. Allow the agarose to cool and prepare the gel tray and casting chamber.
3. Pour the agarose into the gel tray and place the comb into the gel.
4. Let the gel sit and solidify.
5. Prepare samples for loading by adding dye to the sample DNA in a ratio of 1:5.
6. Pull the gel tray out of the casting tray and place it into the gel rig.
7. Fully submerge the gel with 1X TBE and remove the comb.
8. Load the samples into the gel.
9. Run the gel at 130 V until the dye has reached the end of the gel.
10. Once the gel has finished running, stop the power supply.
11. Place the gel on a UV transluminator and using a razor, extract the band of interest.
Column Protocol
1. Following electrophoresis, excise DNA band from gel and place gel slice in a 1.5 mL microcentrifuge tube.
2. Add 10 µL Membrane Binding Solution per 10 mg of gel slice. Vortex and incubate at 50–65°C until gel slice is completely dissolved.
3. Add an equal volume of Membrane Binding Solution to the PCR amplification.
4. Insert SV Minicolumn into Collection Tube.
5. Transfer dissolved gel mixture or prepared PCR product to the Minicolumn assembly. Incubate at room temperature for 1 minute.
6. Centrifuge at 16,000 x g for 1 minute. Discard flowthrough and reinsert Minicolumn into Collection Tube.
7. Add 700 µl Membrane Wash Solution (ethanol added). Centrifuge at 16,000 x g for 1 minute. Discard flowthrough and reinsert Minicolumn into Collection Tube.
8. Repeat Step 4 with 500 µL Membrane Wash Solution. Centrifuge at 16,000 x g for 5 minutes.
9. Empty the Collection Tube and recentrifuge the column assembly for 1 minute with the microcentrifuge lid open to allow evaporation of any residual ethanol.
10. Carefully transfer Minicolumn to a clean microcentrifuge tube.
11. Add 50 µL of Nuclease-Free Water to the Minicolumn. Incubate at room temperature for 1 minute. Centrifuge at 16,000 x g for 1 minute.
12. Discard Minicolumn and store DNA in –20°C fridge.
TA Cloning Protocol
Use the formula below to estimate the amount of PCR product needed to ligate with 50 ng (20 fmoles) of pCR™2.1 vector:
x ng PCR product = (y bp PCR product)(50 ng pCR™2.1 vector)
Where x ng is the amount of PCR product of y base pairs to be ligated for a 1:1 (vector:insert) molar ratio.
1. Centrifuge one vial of pCR™2.1 to collect all the liquid in the bottom of the vial.
2. Determine the volume of PCR sample needed to reach the required amount of PCR product. Use diH2O to dilute the PCR sample if necessary.
3. Set up the 10 µL ligation reaction as follows:
| μL | Reagent |
|---|---|
| x | fresh PCR product |
| 2 | 5X T4 DNA Ligase Reaction Buffer |
| 2 | pCR™2.1 vector (25 ng/µL) |
| 1 | ExpressLink™ T4 DNA Ligase (5 units) |
| 5 - x | diH2O |
| 10 | Total |
4. Incubate the ligation reaction at room temperature for a minimum of 15 minutes.
5. Centrifuge vials containing the ligation reactions briefly and place them on ice.
6. Thaw, on ice, one 50 µL vial of frozen One Shot® Competent Cells for each transformation.
7. Pipet 2 µL of each ligation reaction directly into the vial of competent cells and mix by stirring gently with the pipette tip.
8. Incubate the vials on ice for 30 minutes. Store the remaining ligation mixtures at −20°C.
9. Heat shock the cells for 30 seconds at 42°C without shaking. Immediately transfer the vials to ice.
10. Add 250 µL of room temperature S.O.C. medium to each vial.
11. Shake the vials horizontally at 37°C for 1 hour at 225 rpm in a shaking incubator.
12. Spread 10–200 µL from each transformation vial on LB agar plates containing X-Gal and 50 µg/mL of kanamycin or 100 µg/mL ampicillin.
13. Incubate plates overnight at 37ºC. Transfer plates to 4°C for 2–3 hours to allow for proper color development.
14. Inoculate liquid culture with white colonies for miniprep and sequence confirmation.
Miniprep
1. Resuspend pelleted bacterial cells in 250 μl Buffer P1 and transfer to a microcentrifuge tube.
2. Add 250 μL Buffer P2 and mix thoroughly by inverting the tube 4–6 times for no more than 5 minutes.
3. Add 350 μL Buffer N3 and mix immediately and thoroughly by inverting the tube 4–6 times.
4. Centrifuge for 10 min at 13,000 rpm in the microcentrifuge.
5. Apply the supernatants from step 4 to the QIAprep spin column by decanting or pipetting.
6. Centrifuge for 30–60 seconds. Discard flow-through.
7. Wash the QIAprep spin column by adding 0.75 mL Buffer PE and centrifuging for 30–60 seconds.
9. Discard the flow-through, and centrifuge for an additional 1 min.
10. Place the QIAprep column in a clean 1.5 ml microcentrifuge tube. To elute DNA, add 50 μl Buffer EB or water to the center of each QIAprep spin column, let stand for 1 minute, and centrifuge for 1 minute.
Sanger sequencing
Sanger sequencing was used to confirm MDM2 construct sequence. All sequencing was performed by the UT core facilities.
mRNA Template Preparation
PCR linear template out of PCR2.1 vector
| μL | Conc. | Reagent |
|---|---|---|
| 10 | 10X | PCR buffer |
| 5 | 4 mM | dNTPs |
| 2 | 20 μM | forward primer |
| 2 | 20 μM | reverse primer |
| 2 | 10 ng | template vector |
| 1 | Taq DNA polymerase | |
| 77 | diH2O | |
| 100 | Total |
| Step # | Temp. (°C) | Time (sec) |
|---|---|---|
| 1 | 95 | 120 |
| 2 | 95 | 15 |
| 3 | 47 | 30 |
| 4 | 72 | 60 |
| 5 | Go to step #2 24 times. | |
| 6 | 4 | Forever |
Purify completed PCR on 2% NuSieve agarose gel and recover with Promega Wizard SV Kit.
Run off transcription
| μL | Conc. | Reagent |
|---|---|---|
| 2 | 10X | transcription buffer |
| 2 | 100 mM | DTT |
| 1.5 x 4 | 100 mM | ATP/CTP/UTP/GTP |
| x | 1 µg | dsDNA template |
| 2 | T7 RNA polymerase | |
| 8 - x | diH2O | |
| 20 | Total |
Incubate overnight at 37°C. Then add 1 µL DNase to the solution. Incubate at 37°C for 15 minutes. Add 21 µL of 2X Blue denaturing dye to the solution. Incubate the solution at 65°C for 3 minutes.
ssDNA Template Preparation
PCR linear template out of PCR2.1 vector
To create a template ssDNA, PCR is ran on the newly amplified dsDNA with one primer concentration being significantly higher than the primer in the opposite direction. To increase yield, this reaction was run 8 times and the products were concentrated.
| μL | Conc. | Reagent |
|---|---|---|
| 10 | 10X | PCR buffer |
| 5 | 4 mM | dNTPs |
| 2 | 20 μM | forward primer |
| 2 | 0.2 μM | reverse primer |
| 2 | dsDNA template | |
| 2 | Taq DNA polymerase | |
| 77 | diH2O | |
| 100 | Total |
| Step # | Temp. (°C) | Time (sec) |
|---|---|---|
| 1 | 95 | 120 |
| 2 | 95 | 30 |
| 3 | 45 | 30 |
| 4 | 72 | 60 |
| 5 | Go to step #2 49 times. | |
| 6 | 4 | Forever |
Precipitate DNA product by ethanol precipitation and purify on 2% NuSieve agarose gel. Recover the band below the dsDNA template and process with Promega Wizard SV Kit.
CHA Circuit Testing
Standard curve prep
Measure the fluorescence produced by the Fluorophore at different concentrations. Prepare 0nM, 10nM, 20nM, 30nM, 40nM, 50nM solutions of the fluorophore in triplicate. Load onto a microplate and allow the Safire to take fluorescence measurements for 20 cycles.
Linear catalyst test
Solution Preparation
Prepare an 800nM Reporter solution by mixing equal concentrations of Fluorophore and Quencher strands and diluting in 2x and 1x TNaK buffer solution. Dilute Hairpin1 to 300nM, Hairpin2 to 200nM, Linear catalyst to 200nM using 2x and 1x TNaK buffer solution.
Refolding of Oligos
Refold the solutions in the thermocycler under these conditions:
95°C for 5 minutes.
Decrease temperature at a rate of 0.1°C per second.
Hold at final temperature of 37°C.
Mixing Reactions
Each reaction will be prepared in triplicate as follows, where the amount X is determined by the amount of linear catalyst needed to reach the test concentrations:
| μL | Conc. | Reagent |
|---|---|---|
| 5 | 800 nM | Reporter |
| 5 | 300 nM | Hairpin 1 |
| 5 | 200 nM | Hairpin 2 |
| x | 200 nM | Linear Catalyst |
| 1 | 100 mM | MgCl2 |
| 5 - x | 1x | TNaK |
| 21 | Total |
Because the reaction is initiated upon the addition of Hairpin 1, Hairpin 1 is added into the reaction last to avoid premature initiation of CHA. Mix each of the reactions into a microplate using a multichannel pipette. When adding Hairpin 1, pipette up and down slowly to mix the reaction before loading the microplate into the Safire machine.
Safire Reading/Incubation
Safire is a monochromator-based microplate detection system that has a range of high speed detection techniques. A protocol was set up on the system that would measure and record the intensity of fluorescence. The device provided us with the measurement and record of fluorescence intensity produced by the reaction while keeping the temperature steady at 37°C.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}