User talk:Filip Zembowicz
End of Class Summary
- While I haven't written much on the wiki, I am documenting some of the things that I have accomplished over the course of the semester. I was part of the infrastructure group, and most of my work consisted of coding things for the [filip.freelogy.org] node. I made some contributions to the InfrastructurePage. I was originally planning to wrap up a few things over this weekend, but since the deadline got pushed up to Friday, I am writing my documentation now (and taking a final exam tomorrow... haha) There are some major achievements we made this semester, and here are my thoughts:
CodeIgniter
This was the PHP framework that the trait-o-matic front end operates on. It had its advantages and disadvantages. One major advantage was the fact that it isolated apart different components of the application, so that we could work on the data and presentation separately. This Model -> View Controller -> View paradigm (which is very common in Mac programming, actually) is quite useful because the same designs can be reused over and over. Another advantage of CodeIgniter was the many helper functions -- for example, database access is pretty much almost trivial -- returning an entire set of results was just one PHP line, thanks to CI's helper functions.
At the same time, some of these things made it much harder to work with. For example, the learning curve was a little steep, due to the nature of the system and the very personal nature of Xiaodi's code -- sometimes it wasn't commented well and it took a while to understand what was going on. Another disadvantage was the "babysitting" that CI sometimes feels like it's doing -- for example, by disabling $_GET variables in PHP -- the ubiquitous URL modifiers that make passing variables between different pages a breeze. Overall, I would consider CI if ever doing another data-driven project like this (due to the helpers) but would otherwise pass on using such a system for web development.
The model paradigm
After talking with the modeling and biology groups, we settled on a schema where individual users would be able to submit models based on literature, in order to add functionality to the application without requiring massive advances in the amount of Trait-o-matic stored data in order to parametrize a general model. While Brett had some good ideas about using HTML forms to parametrize models, I thought it would be a good idea to give the end user much more flexibility than that -- so together with Joe, Alex Ratner, and input from the biology and modeling groups, we decided that python would be a good language, given its strong mathematical and statistical packages, should they be necessary for a model, and also the fact that many people are already familiar with python. A good reason to keep it as files and not as just database-residing scripts is that files are sharable, downloadable, and can themselves contain their parameters. In this vein, we created a format wherein all of the necessary information for a model resides within the comments at the top of the python file, in a standardized way. Right now, these include things like rsids of interest, references, and author, but eventually this could also take phenotypic inputs, to create more well-rounded models. These files are uploadable on the models tab (with a password), where they can also be viewed, along with their references. Eventually, the models tab will be a portal for learning about the different models and discussing them.
Hypothesis Page
Creating this model paradigm, I thought it would only be appropriate to create a framework for people to create their own models with, in a simple way. In this vein, I created the hypothesis page, which, using an autosuggest box, allows the end user to query for multiple people's phenotypes at particular rsids (by pressing FIND VARIATIONS), as well as to create "bare bones" python code that can then be supplemented with whatever theories one has about the interactions of genes (or from literature). Hopefully, this will make it easier for people to get started (by pressing GENERATE PYTHON). There is also an option to filter out people, as suggested by Ridhi, since people might want to perform subgroup analyses, but this isn't functional right now. This should be based more on phenotypic data rather than just names, and that just hasn't been implemented yet, but will come soon.
The new results page
This is really a lot of Ratner's work -- but I got the python files to actually execute and create risk assessments based on the models. They are now displayed along with the results for each person. Fortunately, none of our test subjects have abnormal results for these tests, so none of the results differ from each other.
Semantic Categorization
I had initially thought this would be a simple thing -- standardizing the Trait-o-matic phenotypes a bit so that they would get categorized into distinct disease groupings rather than appearing all together. I started off by attempting to use the MeSH classification, however, this was a little to restrictive because a certain disease had a particular way of being referenced, and other semantically similar names were not present, for example, the difference between Type II Diabetes, and Non-Insulin Dependent Diabetes. I then discovered the UMLS Knowledge Source Server [[1]] which is a godsend with respect to semantic classification -- it is a compilation of many different databases on medical and related knowledge, and any single concept can be searched for by any of its names, no matter how obscure, and even in foreign languages :). I still encountered some problems -- for example, in standardizing the search queries, or in finding the most applicable result when multiple results were produced. It also took me some time to understand the UMLSKS web service- it's an archaic Java-based tool, so I had to engineer the service to work with PHP.
So, I achieved a 60% reduction in the amount of categories that the database entries belonged to. While this is significant, it still lacks the precision I thought it needed to be debuted on the trait-o-matic website. For example, many diseases with regionally-associated alleles showed up as separate entries, because they rightfully are in the UMLSKS server. Thus, making a call about whether a semantic call should be observed or more simple matching heuristics should be used was a difficult one in certain cases -- and I wanted this tool to be quite general and not just a collection of solutions to corner cases. In conclusion, with semantic categorization, I learned of many difficulties of natural language processing, and although I am on the way to creating a categorization system, it is still a little far away.
Remaining issues
- Sharing of models -- I think it would be nice if just like with the genomic data itself, the models could be shared by T-O-M nodes on the data warehouse. Then, TOM would be quite useful for some clinicians. For example, if clinicians loaded their local trait-o-matic servers with the models that they were interested in (or, specialists could load the models that they are interested in, such as all models dealing with cancers for oncologists), it would be a great system rather than the current necessity to upload them individually. Such a distribution system would be great too because it could incorporate some of the social aspects of science, such as commenting on research and discussion
- RestrictedPython -- this will be changed really soon (right now uploading is password protected). Sandboxing python will allow for increased security.
- Semantic categorization, as noted above
- Interfacing with parametrizing models:Right now, the modeling group's general models are designed to work with data provided from studies, but it would be great to begin to create infrastructure to allow the model to itself be parametrized from trait-o-matic data. Right now there are just too few genomes for this to be useful, given Frankel's calculations. We could envision a temporary solution -- perhaps loading in the studies' results into TOM in some standardized format might give us "filler" data until more genomes are sequenced. Regardless, allowing the SciPy scripts to query TOM for large arrays of genotypes, in a standardized way, would lead towards the eventual goal of a learning model
- Multi-genome relationships: It would be great to create tools that could be used with families -- for example, tracking the inheritance of particular alleles as being either paternal or maternal. This has an impact on eventual disease susceptibility, and it would be great to parametrize. Another playful tool would be the Cupid idea from the beginning of class - seeing what possible offspring two people would have.
- Renaming TOM -- it might be better off with a more professional-sounding name.
Technical Changes
This is more for my own memory (and to move towards eventually merging our developments into the standard trait-o-matic in the repository).
New tables: Ariel.autosuggest -- contains autosuggest terms in the future these should be loaded from all data sources, not only those present in samples Ariel.files -- contains information about uploaded models
New CI models: File -- made by ratner, contains information about a python file
New views: comparemulti.php -- displays many people's phenotypes together hypothesis.php -- creates hypotheses models.php -- allows for the upload of new models, displays the current models results2.php -- compresses all phenotypic information into one table, displays rsids, outputs model information samples2.php -- allows for searching by chromosomal location now slightly defunct, we are going by rsid
New Controllers: controllers for all above plus modelgen.php -- creates barebones python models for people suggestJSON.php -- handles autosuggest, creates JSON
New Scripts: suggest.js -- does the autosuggest and many other functions of the hypothesis page.
Latests thoughts on computational biology paradigm
Attached is a PDF containing what I have been thinking about in the scope of the project:
File:Biophysics101-fils-discussion2.pdf
Here are the papers I reference:
- The Automation Of Science -> File:The Automation of Science.pdf
- A Query Language for Biological Networks -> File:A query language for biological networks.pdf
Assignment #1
- A3 =k*A2 can be rewritten as [math]\displaystyle{ \frac{dx}{dt}=k*x }[/math] which, when solved for x, gives [math]\displaystyle{ x(t)=e^{(kt)} }[/math]
- plotting this for various values of k gives the following graphic (using python and photoshop for the animation):

- A3 =k*A2*(1-A2) can be rewritten as [math]\displaystyle{ \frac{dx}{dt}=k*x*(1-x) }[/math] which, when solved for x, gives [math]\displaystyle{ x(t)=\frac{e^{(kt)}}{1+e^{(kt)}} }[/math]
- plotting this for various values of k gives the following graphic:

- Plotting with python was easy-- it is clear that the exponential function grows without bounds while the logistic function approaches an upper limit. The logistic function could be something such as found in Michaelis-Menten kinetic modeling-- changing the parameter could represent altering the sensitivity of a promoter or the sensitivity of a switch. However, there is no lower threshold as there is in the family of Hill functions. The exponential function, on the other hand, grows without bounds-- this is something that is difficult to imagine directly in biology, since there are always environmental constraints, meaning that growth of populations may be better modeled by logistic equations rather than exponential ones.
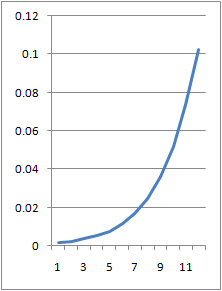
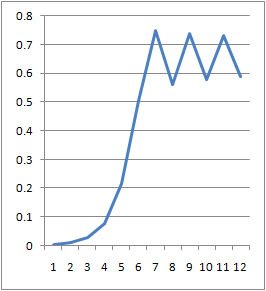
- commenting on excel versus python: python was much nicer to use. Excel required the copying and pasting of formulas, while scripting with python was quick. Python could additionally be automated to consider many different parameter values simultaneously, which would aid in more advanced modeling tasks. However, excel is very simple to learn. Excel also showed some interesting chaotic motion at the top, it runs into some issues when reaching the limit of the equation (as seen above with k=1.5 on the left and k=3 on the right).


Assignment #2
Interesting conclusions: As the length of the peptide increases, the more likely that truncations occur, while the probability of peptide elongation decreases. Some data:
- Peptide of length 8 (offset of +1)
Rate of mutation: 1.0 % Rounds of mutations: 1 Number of trials: 100000 Percentage mistranslated overall, 17.769 Percentage truncated, 0.969 Percentage elongated, 2.551
- Peptide of length 101 (offset of +3)
Rate of mutation: 1.0 % Rounds of mutations: 1 Number of trials: 1000 Percentage mistranslated overall, 90.7 Percentage truncated, 11.8 Percentage elongated, 1.9
So, as the length of peptide-coding sequence increases, the chance that mutations occur and lead to mistranslations increases greatly, and truncations will overshadow proteins that are elongated. This is because there there is only a 3/63 chance that a mutation in a non-stop codon triplet will lead to a premature stop codon, while there is a 61/63 chance that a mutation in a stop codon triplet will lead to a non-stop codon.
Comments on coding: it was quite easy to perform all of these string functions. I would like to improve the mutation feature by using a generator rather than a standard function.
To do: Calculate the probability of truncation at an offset n theoretically
My code can be found here: [[2]]
Assignment #3
I spoke at length with Zach about this assignment and general traits desired in human 2.0. One idea that I think permeated everything we talked about was the idea of immediacy, meaning that genomic data could be used to increase the well-being of humans immediately, rather than after waiting for the next generation of offspring. Although genetic pre-modification will undoubtedly improve the outcomes of preventative medicine, it is hard to imagine that emergency medicine will be done away with completely in the future. Thus, the main ideas that I had was using the large amount of genetic trait information to make drug administration smarter.
- Using genomic information to create a holistic metabolic profile (Pharmacogenomics)
The administration of pharmaceuticals is made far more difficult due to the presence of polymorphism within genes important for metabolism, which affect the concentration and therefore activity of drugs over time. In human 2.0, this shouldn't be a problem-- instead of medicines with two vaguely defined dosing schemes, one for adults and one for children-- I believe that it is within our power to create a system by which it can quickly be determined what dose of a particular drug is appropriate for a particular individual given his or her particular metabolic profile.
For a particular drug, a person can be roughly divided into approximately four phenotypes based on the capacities of their metabolic enzymes: poor metabolizers [PMs], intermediate metabolizers [IMs], extensive metabolizers [EMs], and ultrarapid metabolizers [UMs]. Ultrarapid metabolizers have the problem that drugs cannot attain the clinically relevant serum/tissue concentrations, whereas poor metabolizers quickly saturate their metabolic capacities and can accumulate
The cytochrome P450 class of enzymes catalyzes the breakdown of many different xenobiotics through oxidative mechanisms (a review- [[3]] ). There are 74 families of these enzymes that each share over 40% identity, and many more subfamilies that share over 55% identity. There exists a large amount polymorphism among these genes, both in terms of basepair substitutions that affect kinetic properties directly as well as more indirect events such as gene duplication. Many of the short nucleotide differences are listed in SNPedia's[[4]] entry on drug metabolism. There has already been work done on technologies that would allow the parallel sequencing/determination of the polymorphisms within the cyp450 class -- one good example is that of the AmpliChip CYP450 genotyping test (2006, [[5]] ) that uses microarray technology to rapidly sequence 27 different alleles, including 7 gene duplications. However, the spectrum of this test is limited to CYP2D6 and CYP2C19. These are the most and third-most important cyps according to this table[[6]] of the major targets of various p450 enzymes.
However, the cyP450s are not the only enzyme class with activity in drug metabolism -- over 30 classes of such genes exist, as reviewed in ( [[7]] ), both metabolic enzymes such as the p450s, and things such as transport pumps that affect drug disposition throughout the body. Additionally, pharmacokinetics (the actual binding of drugs to their targets) is very dependent on the genetic structure of the target, which in many cases also exhibit a high degree of polymorphism What I envision is creating a tool that will, given a partial or complete genomic sequence, determine as much as possible about the appropriate dosing regime for the largest variety of pharmaceuticals in the body, taking into consideration the various phenotypes associated with changes in the sequences of drug metabolism and disposition
Once we know the metabolic profile, we can also use this information to change it. Returning to the example of cyp450s-- some of these genes have been isolated from various parts of nature that have properties such as the ability to metabolize a far wider range of xenobiotics than is currently possible. For example, the bacteria Rhodococcus ruber has been shown to have interesting properties such as the ability to live solely on a source of polyethylene or even polystyrene ( [[8]] ). At least part of the underlying mechanism of this strange metabolism is due to cyp450s that are particularly evolved for the purpose, such as one self-sufficient monooxygenase that exhibited oxidative activity towards chemicals such as toluene, ethylbenzene, m-xylene, and 7-ethoxycoumarin ([[9]] ). Genetic knowledge of the p450 class can be exploited to generate novel enzymes through DNA family shuffling, as described in ( [[10]] ). Assuming that a method for targeted genetic modification is developed, I envision being able to alter the metabolic profile, for example by introducing metabolizers of environmental toxins and pollutants, thereby reducing the risks faced by humans from xenobiotics.
The work that is anticipated for this project includes: aggregation of many sources of information about polymorphisms and their associated phenotypes as well as drug targets (this information is at [[11]], generation of a model for the contributions of each polymorphism to the pharmacodynamic and pharmacokinetic profile of each drug, and some method to create actual dosing information based on this data. I anticipate that modeling the contribution of various simultaneous polymorphisms to drug metabolism would be the most difficult aspect of this project.
Assignment #4

- People I have talked to: Alex, Alex, Zach
- Goals
The goal of this program is to quickly output the subjects profile in metabolizing a whole panel of pharmaceutical drugs, given a complete (or partial genomic sequence). This is important because it can help in dosing patients appropriately, which can: 1. Hopefully prevent some of the 100,000 deaths due to adverse drug reactions yearly 2. Ensure that patients are getting doses that are appropriate for their particular metabolisms.
- Components
The first component of this project would be to determine in SNPedia the locations of SNPs that affect the genes that are involved in the effectiveness of drugs. This includes traditional metabolic enzymes such as the cytochrome p450 family, as well as drug pumps and the sequences of drug targets, which may impact drug binding efficiency and therefore the drug activity. From this data, the first module of the program would return a list of addresses along with the gene names and consensus nucleotide at those locations, which will then be analyzed in the case of a particular subject's genome.
The next component of the project deals with accessing the subject's genome. Taking the array of addresses provided by the first module, this module would access the individual genome at those locations and return an array containing only those genes that contain mutations. Ideally, this step would be performed through a web-API. This component would return an array containing those genes that have mutations or differences in the particular individual, along with what particular substitution is there present.
Given this data, we can have the program use a lookup table with clinical pharmacology information (extracted from [[12]]) in order to determine which drugs' metabolism would be affected given the mutations encountered in the subject's genome. This module would return the drugs that are affected -- along with the inhibitors that people should avoid in order to prevent drug-drug interactions.
Ideally, this would be presented in an interactive report, which would allow people to see at first glance what drugs have predicted altered metabolisms, but would allow them to look further and see what mutations led to this conclusion. Furthermore, the program could automatically link to GeneTests for the particular genes and to see what laboratories offer genotyping at the locations, in order to confirm that the mutation is due to a true mutation rather than just a sequencing error.
- Division of Labor
The SNPedia processing would be a great individual project -- using python and a webscraper like BeautifulSoup, an individual could automatically populate a mySQL database with information about addresses of interesting SNPs, the associated genes, and the consensus nucleotides. Then, the python module involved would return this data in an array format, in order for another python module to use.
Another similar data scraping task exists for converting the clinical pharmacology tables into a compter-readable format, for use with the analysis module.
A subproject exists in creating a tool to access genomic information at a particular chromosome and address. This would be very useful for the rest of the project since this will allow us to not have to load an entire genome into memory but rather selectively access those places that are of interest to us.
The other modules could also also be designed independently, given that a standardized schema for information sharing is implemented.
- Generalization
The project as drawn out is just a bare-bones application. I envision that it could be expanded to find information on the general profile of all metabolism, which could lead to some interesting conclusions.
- PLEASE COMMENT BELOW!
