RNAtrain:Scientific Background



Noncoding RNAs
What are noncoding RNAs, and why are they so exciting?
RNA (ribonucleic acid) resembles a lot to DNA in structure, with the exception that the sugar is a ribose and that uracil replaces thymine as a base.
We know RNA principally as a mere messenger between DNA and proteins - a piece of DNA, alias a gene, gets copied into a RNA molecule, and the RNA molecule gets in turn translated by the ribosomes into a peptide via the genetic code. And for a long time, it was assumed that this was the main task of RNA, being a delivery boy who copies corresponding chapter of the requested protein from the instruction manual and brings it over to the construction place.
That's quite a vaste of material, when you think of it, because RNA molecules are very versatile and polyvalent: they can fold into many 3D structures, they can have autocatalytic activities (that means that they can themselves cause their own modification like cleavage or conformation changes) and they can interact with plenty of other types of molecules, like proteins, DNA or other RNA molecules.
Over the last decades however, it became clear that RNA molecules are a very large and heterogeneous family of molecules with way more functions than being a simple intermediate between genes and proteins. As a consequence, the genome contains many genes that don't encode proteins but RNA molecules that function on their own.
Noncoding RNAs are divided into two fundamental classes, the small noncoding RNAs, smaller than 200 nucleotides, and the long noncoding RNAs, larger than 200 nucleotides in length. This size limit is quite arbitrary and has no real functional reason. Rather, you need two different techniques of purification to isolate RNAs of less or more than 200 nucleotides.
Small noncoding RNA
The small noncoding RNAs are the best studied type of noncoding RNA. They are further subdivided into plenty of groups, this time according to their function (which makes more sense, admittingly). Here are some examples:
Micro-RNAs (miRNAs):
They negatively regulate gene expression by binding to messenger RNA molecules, leading either to their destruction or the blocking of their translation into peptides.
Small nucleolar RNAs (snoRNAs):
They guide the enzymes who carry out chemical transformations of the ribosomal RNA to their appropriate location.
Piwi-interacting RNAs (piRNAs):
They defend germ cells against foreign or harmful DNA pieces.
Long noncoding RNA
They are pretty popular right now. The main reason is that it was discovered only over the last years that there are a lot of them, in the sense that there is apparently a lot of DNA previously thought to be silent that is in reality transcribed into RNA molecules. However, per noncoding gene, there are relatively few RNA molecules produced, this is why only the latest, sensitive techniques, able to sequence all RNA molecules of a given cell type, could detect them.
Now it is even speculated, that there are more genes encoding long noncoding RNAs in the human genome than genes coding for peptides. It is still unknown if they all have a function or if most of them are rather transcriptional noise, meaning that the transcription machinery copies some meaningless DNA by error.
Nevertheless, many long noncoding RNAs have been proven to play important roles in virtually all cellular processes:
Some act as scaffolds, in order to connect various molecules in precise places in the cells. Others, through their ability to bind molecules of different natures, attract proteins to specific DNA motifs.
Autophagy
What is autophagy good for?
If you have not eaten the whole day and you are getting very hungry, your body starts to use its sugar and fat reserves in order to maintain sufficient nutrient supply for body functions. Maintaining a nutrient balance is not only important in whole organisms, but also on the cellular level. In times of insufficient nutrient supply, cells turn on a recycling machinery called autophagy (auto = self, phagy = eating). In a simple picture, the cells form rather big vesicles in their cytoplasm that are called autophagosomes. In some way, these autophagosomes resemble “trash bags” in which the cell collects old protein aggregates, damaged organelles, other cellular debris and even bacteria. In order to disrupt the “trash cargo”, the cell adds a “chemical cocktail” (certain enzymes in an acidic milieu) provided by another vesicle, called the lysosome. After fusion of the autophagosome with the acidic lysosome the “trash cargo” becomes broken down into its components (small peptides, amino acids etc). The recycling circle closes when the components are restored to the cell and thus available for the production of new cell material. Interestingly, this recycling system is not only active during times of starvation. Even under nutrient-rich conditions, there is a basal but lower level of autophagy going on, in order to prevent accumulation of damaged or aged proteins and mitochondria. Such an accumulation can cause severe dysfunction of the cells and it has been associated with a range of human diseases affecting the nervous system, the immune system and cancer. Given the importance of autophagy, it needs tight regulation in order to respond fast and accurate to environmental changes. There are not only many autophagy-specific key proteins known that can up- or down-regulate autophagy, but also some RNA molecules. We know e.g. that small non-coding RNAs (e.g. microRNAs) can block proteins, which would usually turn on autophagy during starvation. We are now interested to investigate whether there are also long non-coding RNAs that can regulate this essential cellular recycling machinery. (Written by Imke Ulken)

Senescence
Any living entity is born, grows, and eventually dies. The process of becoming old is called “senescence”, a word referred either to the aging of the whole organism or to cellular senescence. Because cells are the fundamental building blocks of our body, it is commonly believed that cellular senescence underlies organismal senescence. A cell is defined senescent when it is no longer capable of dividing but still alive and metabolically active. When cells become senescent they change their morphology (enlarge and elongated shape), DNA organization and protein secrection compared to proliferative cells. Senescence happens when cells have reached their maximum lifespan: this is called “replicative senescence” and is mainly due to the telomeres shortening. Telomeres are small segments of DNA at each end of the chromosomes and ensure DNA stability. When cells duplicate their genome, telomeres cannot be entirely copied along with the rest of the DNA, so they get shorter with each new cell division until they shorten to a critical length [1]. However, cells can become senescent prematurely due to different stresses such as DNA damage and activation of genes that can promote cancer (oncogenes). This last type of senescence is called “oncogene-induced senescence” (OIS). Cancer is defined as a large group of diseases that involve abnormal cell growth with the potential to invade other parts of the body. As senescence leads to an irreversible growth arrest, it is considered a barrier against cancer development.
Besides a large number of proteins, also non-coding RNAs molecules can regulate cellular senescence. Our efforts are directed toward the identification of long non-coding RNAs that play a role in OIS and so can be important in preventing cancer development. (Written by Giulia Maglieri, Elena Grossi, Li Li)

Cancer/Neuroblastoma
Cancer is a very complex disease that originates because of uncontrolled cellular events. Cancer cells gain some unfavorable features where they divide indefinitely and don’t respond to programmable cellular death signals; giving rise to different types of tumors. Among different types of cancers, neuroblastoma (NB) is the one that arises from improper development and differentiation of neural crest cells. It is considered as the third most common tumor affecting infants and comprises around 7% of the total tumors observed in children. Neuroblastoma contains many molecular subtypes; including low-risk and high-risk tumors. The low-risk tumors are not associated with metastasis, rather they tend to differentiate into mature cells and respond to chemotherapy. On the other hand, high-risk tumors usually spread into different parts of the body and they show unfavorable clinical outcome due to their aggressive nature.
Neuroblastoma is characterized with several genomic alterations; such as focal amplification (1) of the chromosomal segment 2p24. MYCN is a well-studied oncogene which maps to the amplified region. Genetic amplification of MYCN gene is observed in 25-30% of NBs and is mostly associated with aggressive high-risk tumors. In addition, many long non-coding RNAs (lncRNAs) are co-amplified with MYCN gene and they show a similar pattern of expression.
Recently, the neuroblastoma associated transcript 1 lncRNA (NBAT1) was reported as a risk factor which is differentially expressed between high-risk and low-risk NBs. Less expression of NBAT1 leads to more proliferation of cancer cells, while higher expression level induces cellular differentiation and maturation. Using NBAT1 expression as an independent prognostic marker enables the prediction of overall survival and clinical outcome among different groups.
Systematic investigation of stage-specific as well as subtype-specific lncRNAs will lead to a more comprehensive understanding of NB progression. Also, it may help in revealing more about regulatory networks that govern neuronal differentiation and proliferation. Modulating the expression of important lncRNAs might provide a possible treatment for NB patients. (Written by Mohamad Gendy)

Myogenesis
Skeletal muscle is made up of individual components known as myofibers, which are multinucleated cells formed during muscle development in a process termed myogenesis. While a mammalian embryo is developing, progenitor cells (1) become determined for the myogenic lineage (determination step) and give rise to proliferating myoblasts. Myoblasts will transform to myocytes which withdraw from the cell cycle, and fuse to one other to form multinucleated myotubes (see figure). Myotubes at their final stage of differentiation will give rise to myofibers. During adult muscle regeneration and growth, myoblasts are derived from resident muscle precursor cells (3), termed satellite cells. Satellite cells are mitotically quiescent (2), and they reside in muscle tissues in association with myofibers. Upon a growth stimulus or injury, satellite cells activate, proliferate and undergo differentiation process to produce new myofibers. Myogenesis is a highly organized process, many transcription factors (4) are known to be important for regulation of different steps of this procedure (see figure). miRNAs (5) and lncRNAs are another factors which recently have been discovered as myogenesis regulators. (Written by Sama Shamloo)

Glossery: (1) A progenitor cell is a biological cell that, like a stem cell, has a tendency to differentiate into a specific type of cell, but is already more specific than a stem cell and is pushed to differentiate into its "target" cell.) (2) Quiescent cell is neither dividing nor preparing to divide. (3) Precursor cells are stem cells that have developed to the stage where they are committed to forming a particular type of cell. (4) Transcription factor is a protein that binds to specific DNA sequences, thereby controlling the rate of transcription of genetic information from DNA to RNA. (5) A microRNA is a small non-coding RNA molecule found in plants, animals, and some viruses, which functions in RNA silencing and post-transcriptional regulation of gene expression.
Oocyte to Embryo Transition
Oocyte to zygote transition (OZT) is the most important transitions in life. OZT refers to transformation of a differentiated oocyte into a zygote, which has potential to develop into a new organism – i.e. zygotic cells acquire totipotency, which refers to their ability to give rise to any embryonic and extra embryonic cell types. Oocytes start to form already during embryonic development when oocyte precursors (primordial germ cells) migrate into the future gonad. In mice and humans, oocytes are already formed at birth and rest in the ovary. Some of the oocytes subsequently enter growth phase in response to periodical hormonal stimulation in sexually mature females. During the growth phase, oocytes grow in size and their genes synthesize factors, which will support future development. Factors stored include a wide range of RNA molecules. Fully-grown oocytes stop their transcription and are released from the ovary for fertilization. Upon fertilization, a massive reprogramming of gene expression takes place, which transforms a differentiated oocyte into the totipotent zygote. This reprogramming is driven by maternal factors stored in the oocyte until the control is taken over by zygotic gene expression. Therefore both maternal and zygotic factors contribute to this complex transition. In mice, the major phase of zygotic genome activation takes place at the 2-cell stage while in other mammals usually occurs a bit later (4-16-cell stages). Although several studies have been performed to unveil the mystery of OZT, key factors involved are yet to be identified. Our lab studies contribution of non-coding RNA to the genome reprogramming during OZT in mice. (Written by Sravya Ganesh)
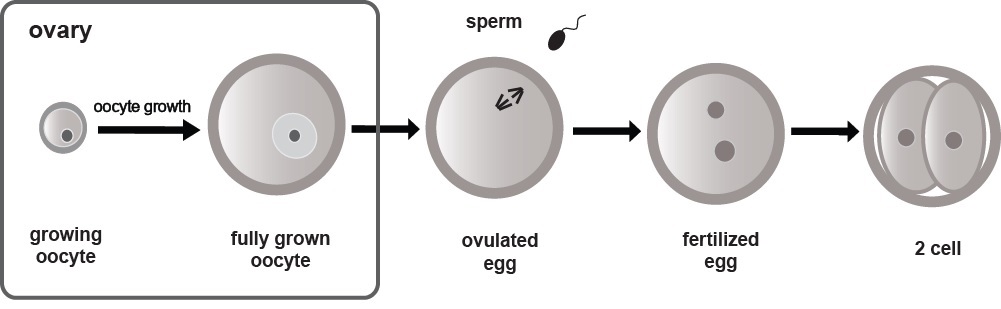
.jpg)
Colorectal Cancer
If you daily consume high intake of fat, alcohol, red meat, smoke and lack of physical exercise, you may belong to a group of high risk of colorectal cancer (CRC). CRC originates from the epithelial cells lining in the colon or rectum, some of which have abnormal growth activity and ability to invade or spread to other parts of the body. Currently, scientists are putting high effort to find the genes or signaling pathways which lead normal cells to become cancerous with the hope to early prevent the tumor development and metastasis. Genome-scale analysis and next generation sequencing has revealed that many protein-coding genes and non-protein-coding genes are all involved in causing abnormal colorectal cancer cells.
These abnormal cells are most frequently as a result of mutations, which can be inherited or acquired, in the Wnt-signaling pathway. APC gene is the most commonly mutated gene in all colorectal cancer. The APC gene produces APC protein, which prevents the accumulation of β-catenin protein. When β-catenin accumulates, they translocates (moves) into the nucleus, binds to DNA, and activates the transcription of proto-oncogenes. Other mutations occur in cancer-related genes, such as p53 or BAX.
With the advantages of next generation sequencing (refer to RNA-seq from Foivos Gypas), there are more and more evidence indicating that non-coding RNAs, either small non-coding RNAs or long non-coding RNAs, are frequently aberrantly expressed in cancers, and some of them have been implicated in CRC development and metastasis. Further characterization of these non-coding RNAs will not only give us new insights into the molecular biology of CRC development but also provide us potential non-coding RNA therapeutic prevention in the future. (Written by Hung Ho)
Development of siRNA pools
Due to the rapidly expanding role of RNA in human health and disease, scientists are developing methods to specifically regulate different RNAs to understand their function. siRNA or silencing RNA was discovered in the 1990s by Craig Mello and Andrew Fire, for which they were awarded the Nobel Prize in Physiology/Medicine in 2006. They found that introducing double-stranded RNA into worms (C. Elegans) reduced the expression of certain genes. It was later revealed that on entering the cell, double-stranded RNA is cleaved into smaller lengths of ~21-25 ribonucleotides by an enzyme called Dicer. These small intermediates carry out the gene knockdown effect, specifically the antisense strand would bind to a complementary RNA sequence from an endogenous target gene, recruiting it to a protein complex called RISC or RNA-induced silencing complex. This complex contains Argonaute proteins which then carry out the “slicing” of the target gene, hence silencing its expression. This heralded a burst in siRNA research as it provided a simple and fast way to regulate gene expression. However, it slowly became clear that these siRNAs had various off-target effects (i.e. that one siRNA can bind specidically to several mRNAs, resulting in lower specificity of the knockdown). siPOOLs were therefore developed by siTOOLs Biotech GmbH. siPOOLs are complex pools of accurately defined siRNAs and dramatically reduce off-target effects while producing robust target gene knock-down. They therefore deliver clean and reliable phenotypic data. My work focusses on improving the development of siPOOLs and developing siPOOLs against non-coding RNA. (Written by Catherine Goh)
Cellular stress response
Cells in human body are constantly facing threats to their wellbeing. Extremes of temperatures, exposure to toxins, radiation and mechanical damage are potentially harmful and in order to survive cells have to adapt to them. Molecular changes that cells undergo in response to those environmental factors are generally referred to as cellular stress responses. Depending on nature and severity of the stress cells may survive exposure to it or be killed by it. If the stress is mild or cells are well suited to cope with it they survive and their biology remains unaltered. If the stress is too severe for the cells to handle they may be damaged and their biology may be altered. Those alteration in most cases lead to cells death. In some cases however cells survive and the alterations induced by the stress conditions may lead to disease. Stressors with potential to induce genetic mutations are particularly dangerous for human organism and may lead to serious diseases including cancer. In order to better understand diseases that are connected to cellular stress cellular stress responses have been intensively studied. Those studies resulted in identification of many molecules and mechanisms regulating the stress responses. However our understanding of the subject is far from complete. Protein driven regulation of cellular stress responses have been most intensively studied and is best understood. However new lines of evidence point to long non-coding RNAs as potentially critical components of cellular stress responses. We are now trying to identifying long non-coding RNAs involved in cellular stress responses and discover their precise functions. (Written by Karol Rogowski)
RNA-seq
RNA-seq or RNA sequencing is a recently developed technology based on next-generation sequencing that is rapidly replacing the older microarray technology. RNA-seq has a broad range of applications, but one of the most important is to detect and quantify specific RNAs (coding or non coding) at a given moment in time. An RNA-seq experiment can be devided in two parts. The first part is the experimental, where samples are prepared and sequenced and the second part is the computational where data are analyzed. In order to perform an RNA-seq experiment (see figure below), the first thing one should do is to extract the RNA samples. The samples are then cut into small pieces (RNA fragments) and reverse transcribed and amplified in order to generate multiple cDNA fragments. Then these fragments are inserted into a next generation sequencer that generates reads. Reads are the sequences of the small fragments. After this step data are analyzed computationally, since the reads is actually a big text file. In order to find which transcripts or genes are expressed you need to align the reads to the genome (or the transcriptome). A good program to align reads is segemehl. Aligning reads is a computationally expensive task, so your computer should be supplied with sufficient amount of memory, processing power and disk space. You can see the results of the alignment using a genome browser. A nice solution is IGV. Apart from viewing results you can also quantify, the expression level of genes or transcripts. This can be done either by counting how many reads map to a specific location, or by using some more advanced probabilistic methods. Some of these methods can be found here.
(Written by Foivos Gypas)

Techniques to visualize RNA
Understanding the mechanisms by which lncRNAs function in cells can be facilitated by knowing their subcellular localization. In situ hybridization (ISH) technologies are powerful methods to visualize RNA molecules within cells and tissues. ISH also allows the relative quantification of the RNA abundance in different cellular compartments or cell populations within a tissue. In the last years, diverse technologies have emerged with significantly increased sensitivity and specificity in RNA in situ detection. These different technologies differ in their probe types and on their detection principle. The most common and well-established commercially available probe types are (by alphabetic order): • Branched DNA Technology – it is based on up to 20 probe pairs per target RNA type. The detection system generates a “tree” of fluorophores, amplifying the signal more than 8000x (see http://www.acdbio.com/products/rnascope-assays and https://www.panomics.com/products/rna-in-situ-analysis/view-rna-overview) ; • LNA-based Technology – probes contain LNA-modified nucleotides that confer higher stability and specificity of the probes by increasing their hybridization properties. The detection of target RNAs usually requires enzymatic amplification (see http://www.exiqon.com/microrna-in-situ-Hybridization); • Stellaris FISH Technology – uses up to 48 DNA labelled-probes to the target RNA and relies on a direct visualization of the signal (see https://www.biosearchtech.com/display.aspx?pageid=222); Obviously, different probe types, and associated detection methods, have different sensitivity and resolution capacity. It is therefore essential to understand the advantages and limitations of each probe technology and adapt it to the characteristics of the RNA/miRNA/lncRNA under study. In this project, we are interested in determining and improving the best in situ hybridization methods to visualize lncRNAs. We are also interested in combining in situ hybridization with other methods to simultaneously detect other types of molecules, such as DNA or proteins. Finally, we intend to establish methodologies to visualize and document, in situ, the close proximity, and possibly, the interaction, between two different molecules. (Written by Ricardo Soares)
