OhioMod2013:Team/Paul
<html>
<head> <style type='text/css'>
.firstHeading, #column-one, #p-bookmarks, #p-history , .portlet{ display:none; }
- bodyContent, #content{
display:none; background-color:#FFFFFF;
}
- column-content, .container, #globalWrapper, body{
background-color:#FFFFFF; }
p{ color:dimgrey; text-align:center; margin-left:auto; margin-right:auto; font-gamily:Calibri; font-size: 15px; }
.selflink{ display: table-cell;
text-decoration: none; font-family: Gill Sans MT; outline: none; font-size: 20px; color: dimgrey; border-right: 2px solid dimgray; padding: 15px 25px; /* sets button size */ font-weight:normal;
}
ul.navigation-bar {
list-style-type: none; border: 2px solid dimgray; padding: 0px; margin-left: 20px;
width:615px;
*line-height: 3.2em; /* IE6,IE7 */ }
ul.navigation-bar li {
display: table-cell;
background-image: -webkit-linear-gradient(bottom, silver, azure); background-image: -moz-linear-gradient(bottom, silver, azure); background-image: -ms-linear-gradient(bottom, silver, azure); background-image: linear-gradient(bottom, silver, azure); background-color: midnightblue; /* IE6...IE9 */ *display: inline; /* IE6,IE7 */
}
ul.navigation-bar li:hover {
background-image: -webkit-linear-gradient(top, silver, azure); background-image: -moz-linear-gradient(top, silver, azure); background-image: -ms-linear-gradient(top, silver, azure); background-image: linear-gradient(top, silver, azure);
}
ul.navigation-bar li a, ul.navigation-bar li ul li a {
display: table-cell; text-decoration: none; font-family: Gill Sans MT; outline: none; font-size: 20px; color: dimgrey; border-right: 0px; padding: 15px 25px; /* sets button size */ }
ul.navigation-bar li ul{ list-style: none; display: none; position: absolute; padding: 0px; margin: 1px 0px 0px -1px; background-color:#FFFFFF; border: 1px solid dimgrey; z-index:5; } ul.navigation-bar li ul li{ background-image: none; background-color:#FFFFFF; border-right: none; border: 0px solid dimgray; display: block; float: none; position: relative; }
ul.navigation-bar li ul li:hover{ background-image:none; background-color: lightgrey; }
ul.navigation-bar li:hover ul{ list-style: none; display: block; position: absolute; }
ul.navigation-bar li a{ display: block; }
.profile-right{ clear:both; }
.profile-right p{ text-align:left; float: left; } .profile-right img{ float: right; padding: 20px; }
.profile-left{ clear:both; }
.profile-left p{ text-align:left; float:left; }
.profile-left img{ float: left; padding:20px; }
- OSUfooter{
clear:both; }
- protocol-content table td{
border: 1px solid black; text-align:center; }
- protocol-content p{
text-align:left; }
.overlay-bg{ display:none; position:fixed; top:0; left:0; height:100%; width:100%; cursor: pointer; background: #000; background: rgba(0,0,0,0.75); }
.overlay-content{ background:#fff; padding:1%; width: 60%; position: relative; top:15%; left:50%; margin: 0 0 0 -30%; cursor: default; -webkit-border-radius: 5px; -moz-border-radius: 5px; border-radius: 5px; box-shadow: 0 0 5px rgba(0,0,0,0.9);
}
.close-btn{ float:right; font-size:x-large; margin-top:-10px; }
</style>
<script src="//ajax.googleapis.com/ajax/libs/jquery/1.10.1/jquery.min.js"></script>
<script type="text/javascript"> $(document).ready(function() {
$('.show-popup').click(function(event){
event.preventDefault();
$('.overlay-bg').show();
});
$('.close-btn').click(function(){ $('.overlay-bg').hide(); });
$('.overlay-bg').click(function(){
$('.overlay-bg').hide();
});
$('.overlay-content').click(function(){ return false; });
}); </script>
</head>
<a href="http://openwetware.org/wiki/Biomod/2013"> << Back to Team Pages</a>
<img src="http://openwetware.org/images/1/15/Ohiomod2013.jpg" height="60%" width="60%" style="margin-left:-40px;">
</html>

Paul Gruenbacher

About
Interests
Notebook
Will be using links to google drive for files.
Friday May 10
Submitted cadnano design of 66 helices, non-hollow: 66helicev1. Will note that midline there are scaffold crossovers that do not have staples nearby, had incorrectly assumed that staple crossovers must avoid scaffold crossovers. Design approximately 40 nm long and less than 22 nm in diameter.

Sunday May 12
New version edited by Dr. Castro.66 helices, 2-layered, hollow: 66helicev3. Will note that diameter remains the same, is approximately 44 nm long now. Should not be an issue



'Literature review': It seems well accepted that crossing the nucleolus membrane is the greatest challenge facing gene therapy. The calcium phosphate protocol as it stands only takes us to the cytosol, so the origami must be designed to be able to enter the nucleus. A paper in 1988 used a range of gold particles to measure the size able to enter through the nuclear pore complex [1] and found that the limiting size was 26 nm. This suprised some people as some viral proteins are larger than this, including Hepatitis nucleocapsids. A later paper in 2002 used gold particles with protein complexes to measure a larger size of 32-36nm [2]. We would need to add targeting sequences or nuclear localization signals to the origami. For NLS peptide conjugation to the DNA staples, one would need DNA staples with a free alkylamino group of a thymine. see File:66 helice final v3.json
However, peptide conjugation and staple modification is making things more complicated than what is desired or allowable. Perhaps nuclear targeting sequences would be better, though a recent paper in 2011 downplays their importance [3].
Monday May 13th
Ordered staples for version 3 origami design edited by Castro. The templated had now been decreased to the smaller 7202 template strand. Will be leaving for Puerto Rico while waiting for staples to come in, will be back on the 23rd.
Corresponded with Dr. James Adair at PennU and Dr. Robert 'Bob' Lee of OSU pharma.
Literature Review: In general, some better indications that the origami can be functionalized with cationic peptides to facilitate transport across the nuclear pore complex.
Rosenecker's lab synthesized a tetramer of the PKKKRKV NLSV404 peptide sequnece spaced by glycine residues. (may want to consider finding a lab that has a Applied Biosystems Automatic Synthesizer) [4]. A strong paper, with luciferase transfection assay, in situ flourescent hybridization to detect plasmid dna, and a nuclear import assay with labelled BSA-BODIPY to be transported by the peptide. The SV40 NLS sequence they used is the minimal sequence observed on the T-antigen of SV40 necessary. HeLa cells showed 58.9% transfection within 2 hours, much faster than with the dendrimer alone or with a mutant peptide control. Analysis shows quicker localization of the peptide-plasmid in the nucleus, indicating that the peptides probably helped transport the plasmid across the pore if not through the cytosol as well. Early cell transfection also indicates a lessened need for mitosis to occur. Also did a nuclear transport inhibition protocol by Sebestyen [5], which had also showed linking of peptides to plasmids by the cyclo-propayrroloindole cross-linker. Remember: Karyopherins are the proteins found to accumulate in the nucleus.
Dr. Davis at Case Western also showed that DNA was binded the nucleolin receptor on the cell membrane, and may indicate a new cellular uptake mechanism with increased transfection efficiency. The DNA nanoparticles were formed by PEGlyated polylysine. We probably don't want to pursue this mechanism, but the effectiveness of the internalization and transport of the nanoparticles is impressive [6]. Davis had also demonstrated using PEGylated=polylisine/lipid/DNA nanoparticles that are 10-20nm in diameter and 100 nm in length that are rapidly uptaked and concentrated into the nucleolus. This confirms the aspect ratio hypothesis, and they also seem to enter by nucleolin as well. Davis also mentions compacted DNA being known to increase efficiency so long as the ellipsoidal diameter of the compacted DNA does not approach 25 nm, and this was shown in non-dividing cells[7].
Dr. Dean is one of the first to have described the nuclear targeting sequence of the SV40 DNA itself. In his review [8], Dean describes several methods to nucleari import of nonviral vectors. The classic SV40 NLS PKKKRKV. He also describes the 72 bp sequence that is shown to have binding sites for a number of mammalian transcription factors that are usually brought into the nucleus for RNA transcription. Reich has also idenifitied NFkappaB binding sites that show enhanced expression as well. Using the DNA sequences have an obvious advantage to us in that it eliminates the need for the peptides, but I'm worried that the origami will not allow for the normal binding of the factors even if the binding sequence is spaced and free. Also, we also need to take pore size into account, peptides keep the origami size efficiently small. If we use the peptides, it is important we use a mutant peptide as a negative control. It is also important we check to make sure there is no immunogenic response to the peptide. Dean also mentions the use of the amphiphathic trans-cyclohexane-1,2-diol to disrupt hydrophobic region of central NPC channel. If the DNA origami were to have methyl groups around it, it may help as well.
Side Note: DNA origami would probably be an effective way of DNA labelling, so that only certain regions are exposed for labelling such as biotinylation. Especially if we have a single loop exposed from the origami after it is coated with a protective polymer or such. Then only that loop would be treated with the label.
Friday May 24th
DNA staples arrived two days ago. Pooled the staples into four groups of pre-stock: 96 core staples from plate p086, 41 core staples from plate p085, 13 neighbor staples, and 13 polymer staples. The polymer staples are thus named in the event we want to replace them with staples that will polymerize the origami into filaments.
Created 500nM working stock solution of all staples, and used template strand M13mp18. Final concentrations were 20 nM staples, 10 nM template. The DNA origami was folded under a magnesium screen, ranging from 12, 14, 16, 18, 20, 22, 24, 26 mM. Two groups of 2.5 and 4.5 day ramps were then created. There were 16 groups total.
CalcPhos_origamiv3_orderform_5-24.xls
WorkingStocks_5-24_origamiv3.xls
Monday May 27th
Memorial day, harumph harumph.
- Need to schedule TEM this week, get Nanotech West Access, Order calcium phosphate materials
- Literature notes:
- <300nm is the accepted size for tumor extravasation. keep this in mind for any sort of drug delivery mechanism.
- Zhao Xiaobin makes some good points about FR-targed gene therapy as a target for cancer cells.
- Ward et al. shows that the PEG spacer for the folate is important for efficiency.
- There are groups showing FR-targeting with polyplexes, cationic lipids, PEI. FR-targeted liposomes seem to be more efficient than lipofectim commercial products.
- Concerns: endogenous levels of folic acid and competition, intratumoural diffusion, size, insufficient levels of FR on the tumor cells. Coadministration with anti-estrogen or retinoid receptor ligands which can increase FR expression.
Tuesday May 28th
Found a good protocol for using PEG-bis amine with NHS-activated folate [9]. Scheduled TEM training next week. Called Sigma for their materials, here is a list of what I'm thinking at the moment.
| Name | Cost | Amount | link |
|---|---|---|---|
| Igepal CO520 | 107.00 | ~500 ml | [1] |
| Cyclohexane | 81.60 | 1 litre | [2] |
| Calcium chloride | 29.90 | 100 g | [3] |
| Disodium Phosphate | 26.10 | 100 g | [4] |
| Sodium Citrate | 23.60 | 25 g | [5] |
| Sodium Metasilicate | 31.00 | 25 g | [6] |
I don't think the metasilicate is absolutely necessary, it seems that it is mostly served as a nucleation agent to help the precipitation of the CPNP, though I also thought it helped limit the size of the particles. I do know it is in Adair's protocol, and there is literature of CP-silicate nanocomposites that exist elsewhere, so I think it's safe to go ahead and make sure we have it.
Also note: Motskin shows cytotoxicity of hydroxyapatite nanoparticles with concentrations above 250 μg/mL, which I'm not too worried about especially systemically [10], especially since they were microparticles and they drowned the cells in them, and KEster had shown doped CPNPs had no cytoxicity.
possible flourescence schemes
- Propidium iodide - intercalating flouroescent, excitation 535 nm, emission 617 nm. Good DNA origami marker.
- Dope CPNP with flourescein, lanthanides, or Cy3 amidite (Adair)
- Indocyanine green (ICG) as an infrared flourophore for animal trials (Adair bioconjugation)
- Dr. Castro mentioned YOYO-1 and SYBR Gold. These seem to be ultrasensitive Nucleic acid detection and quantitation, mostly due to their precise proportion of dye to the bases.
Finally, I am still thinking there is a huge potential of origami in DNA engineering, by organizing the DNA origami in such a way that the encoding domains are protected while other non-important regions are exposed to some sort of steric-hindered chemical treatment.
Literature Review:
Nongradative intracellular trafficiking of highly compacted polymeric DNA nanoparticles 2011 by Kim and Nanoparticles of Compacted DNA Transfect Postmitotic Cells 2003 by Liu
Both of these papers are highly suggestive that compacted DNA is a good thing for the gene transfer. Both labs use 30-mer lysine polymers linked to PEG. The cationic lysine condense the DNA into compacted shapes. Liu had shown that compacted DNA up to 25 nm showed good nuclear transfection efficiency, which is limited by the size of the DNA (after 9.9 kb). Liu also suggests that the polylysine incorporated into the condensed DNA binds to alpha importin with equal affinitiy as an extended SV40 large T antigen NLS, which he points to two immunosorbent binding assays as proof[11]. That's quite a statement.
In general, it seems that poly Lysine or pLy can form toroids of 80-100 nm with DNA. This structure depends on a lysine/nucleotide ratio of 0.4. Chan's lab[11] had shown that the polylysine works when coupled with T-ag NLS (P101) works for importin binding and transfection efficiency.
Wednesday May 29th
Don't forget Dr. Jim Lee has his general lab meetings on 4pm Sunday afternoons.
Talked to Adair. Sent me a patent that summarizes much of the work. Funny enough, they are using 200 micron glass beads that one would use in road paint to help make them reflect light at night! So that actually makes the stationary phase of the HPLC a lot cheaper, since these are easier to obtain.
Found HPLC at Dr. Poirier and Dr. Ottesen's labs. I've suggested taking 200 micron glass beads from Polysciences Inc.[7], and packing them into a 5x55 mm column. The glass beads are cheap, and they would seem to be effective by Adair's suggested Van Der Waals chromatography in which the calcium phosphate will bind to the glass beads in a nonionic eluent of ethanol. If this is sound theory, then we're looking at a rather simple HPLC protocol. Need to hear back from Ottesen on how she sets up the HPLC and what sort of spectrophotomoter and such I could be using. 280 nm is the absorbance for Igepal CO-520 apparently.
Lab work
Storage: The 16 groups using 7249 template, 2.5/4.5 days are stored in the fridge in a box with my name labelled on them.
Ran the origami in gel. See OhioMOd2013:Methods/gel electrophoresis for more details.
I stored pieces of gel that contain each band of fully folded origami away in the fridge. Uploaded picture of the gel below. Will note that lane 12 has a band that is slightly raised, that is the one I took out of the gel. All other gel pieces were taken from the second band observed. You'll notice that lanes 3-5 and lanes 12-15 have the best looking bands. There is significant agglomeration to the later bands where there was increased Magnesium concentrations.
Lane 1 DNA 1kb ladder. Lane 2 DNA phage 7249 control. Lanes 3-10 2.5 day folding, 12-26 magnesium conc. from left to right. Lanes 11-18 4.5 day folding, 12-26 magnesium conc. from left to right.
Friday May 31st
Not much to do for Thursday or Friday. Simply waiting on others.
- Maleimide-PEG-NH2 from PEG creative works.[8]
- Microemulsion notes: Igepal CO-520 is a nonionic surfactant. Read The colloidal stability of flouresecent calcium phosphosilicate nanoparticles. This paper is the best justification for using Igepal, and using Citrate as the dispersion. Citrate serves as a highly charged small molecule effective for dispersion. Paper discusses whether the particles will irreversibly agglomerate with drying.
- Read the DLVO theory. DIscusses the distribution of charges in ionic solutions. Also discuses double layers forces to explain phenomena found in the colloidal sciences.
- Use this for DNA mass/molar calculator
- [9]
- So if I use 20 nM template concentration for my folding, with 100 ul final volume, then assuming 14 kb structure size at the end, and assuming 100% folding efficiency, then there is 9.09 ug of origami total, yeech, we can get reporter plasmids for 10 mg. I'd like a 100 μg, but it looks like I'm going to be using a smaller range for loading. Maybe something like 0.1, 0.5, 1, 2 μg for every 1 μg of nanoparticle. I can up the concentration by not bothering with magnesium screen and only use 16 magnesium concentration.
- Since 3x10-3 Molar phosphate concentration and 500x100-3 Molar calcium for the microemulsion method, then phosphate will be a limiting factor by no more than 1:1 molar ratio, unless we get monomer crystallization.
- Should probably first measure mass of calcium phosphate nanoparticles without DNA loading.
Monday 6/3
Still waiting on others. Here's a picture of the nuclear pore complex taken by Dr. Martin Goldberg from Durham University.
Left image: Cytosol side of the membrane. Composed of the cytoplasmic ring, of eight 50 nm long cytoplasmic filaments, with a gap of 70 nm. Right image: Nucleus side of the membrane. Composed of nuclear filaments joining to create a basket-like ring with a gap of 30-50 nm. The central pore itself has a gap of 45-50 nm. The nucleoporin is composed of around 50 different nucleoporins, which occur in multiples of eight.
It's such a good picture I went ahead and made a page simply for the nuclear pore complex.
Tuesday 6/4
Well at least the order was approved, still waiting for delivery for materials.
Composed paper asking donation/collaboration from CLL group. Will probably send to Dr. Byrd with Chris Lucas as cc.
Will continue working on Adobe illustrator and on the website.
Wednesday 6/5
Performed crunch-n-squeeze purification on the gels I had cut last week. Most of the lab does their purification soon after the cutting of the gel, but I see no problem with storing the origami in a gel for over a week. Added to protocol in methods.
Will do TEM grid Uranyl stain with Carl today, added protocol in methods. I stained purified lanes 4 and 5. Its stored in the top left cabinet with my name and information taped. 4 carbon grid was significantly damaged, so check hte periphery for that. lane 5 carbon grid was well done, but since it was the second grid performed there may still be more crystallization. Hopefully all goes well Friday. I just need one picture of the folded structure.
Literature Reivew: Went over Maitra's paper [12] in which she states calcium phosphate can be considered a third generation nanoparticle. Instead of Igepal they used Aerosol OT from sigma, and n-Hexane rather than cyclohexane. A good sign is that she only adds 2.94 μg of pDNA to a 25 ml AOT microemulsion of 70 μL 0.35 M phosphate, which was then added to another 25 ml AOT microemulsion containing 70 μL of 1.36 M calcium solution. They were mixed at 20 drops per minute with continuous stirring at 4 °C for 12 hrs. Then reaction is dissolved in absolute ethanol by vortexing. Solution was pelleted for half an hour at 8000 rpm at 4 °C. The pelleted nanoparticles were washed in ethanol three times, then disperesed in distilled water, and dialyzed overnight using 12 kD dialysis membrane bag.
To determine pDNA encaspulation efficiency, she ran 3 to 9 μg of DNA until distinct DNA bands were observed outside of the nanoparticles. Also ran zeta potential at every pH point. This is a pretty good paper to follow, and I may try to adapt this to Adair's protocols.
Also, I found a paper I hadn't seen before [10] recently published in Jan 2013. They also use the double reverse microemulsion approach using a different non-ionic surfactant ethylene glycol monobutyl ether. There microemulsion was combined for several minutes as well, and citrate was used as the dispersing agent for 10 minutes. THey then centrifuged at 14000 rpm for 15 minutes, and then washed with ethanol and PBS twice to remove residual ethanol. Then dispersed in PBS. Huh, maybe we can do away with HPLC for the time being.
They also measure loading efficiency by agarose gel elecrophoresis and ethidium bromide staining. They loaded with antisense oligonucleotides, so smaller loading. To make sure citrate is on surface, use characterization by the FTIR spectra. USe X-ray Diffraction to determine the phase.
Here's a dissertation paper on nonviral delivery[11]. Random notes from it: HA1, GALA, and melittin are peptides with well-known ph-sensitive endosome lysis. Luciferase is the most reliable indicator of gene transfer. Can use bioluminescent imaging to quantify the luciferase expression in vivo. BLI uses a camera to detect photon emission at 610 nm. There are little false-positive results. I could simply test to see luciferase in the liver.
John Wolff, Professor of Pediatrics and Medical Genetics, U of W-Madison: "Nuclear targeting is the grand problem facing the non-viral gene therapy community". Jesse Gelsinger first to die from adenovirus gene therapy. Can try using H1 linker histone to help wrap the DNA origami? Useful for delivery possibly. See: Peptide nucleic acid staples. Also, photo-chemical coupling of peptide to DNA.
Oh, and the items were finally shipped yesterday!
Thursday 6/6
Waiting on materials. Will try to further prepare materials prior to their arrival.
Friday 6/7
Did TEM images with Carl. Will upload quality pictures shortly. Will probably take some dimensioning as well. Structures seem well-folded with a few structures attaching to one another at the ends. This is possibly due to how the structure was edited by Dr. Castro for the possibility of of being filamentous.
I also started another DNA folding for bulk production. Used the same folding excel sheets as last time on Friday two weeks ago. Will run at 16 mM MgCl2 and 2.5 day thermal ramp. Will use 20 nM scaffold and 50 μL total volume for each PCR tube. Will run two of these sets for 16 total, and 800 μL of around 20 nM origami. This suggests a 78.99 μg yield assuming ~100% folding. For my initial reaction runs next week, I can use a small range of 0.1, 0.5, 1, 2 μg of DNA for 4 different reactions, thus using 14.4 μg.
Here are the images taken. 1st picture forgot to add captions to image. There are some structures that are sitting vertically with the hollow end visible. This is somewhat suprising and is rarely seen in origami structures, so a lucky find!
| 14 mg conc. | 
|
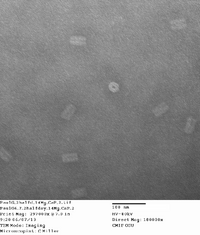
|
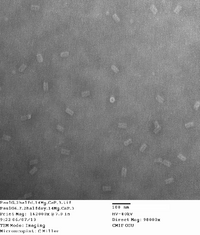
|

|
|---|---|---|---|---|
| 16 mg conc. | 
|
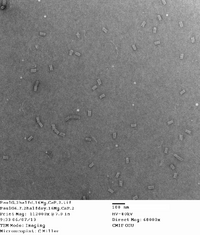
|

|
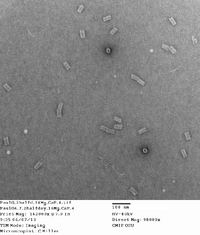
|
Monday 6/10
Finished bulk production of structures, they're sitting in 4°C currently.
Will be meeting with Dr. Poirier with Dr. Castro today, will be discussing use of the HPLC. Currently I feel that I should explore other purification whether by pelleting and evaporation or by other means before I try to adapt to an expensive and complicated HPLC method.
Stock Solutions
| Material | Molar Mass | Measure Out | Solution |
|---|---|---|---|
| CaCl2 | 110.98 g/mol | 0.5549 g in 50 ml | 1x10-1 M |
| HNa2HPO4 | 141.96 g/mol | 0.425 g in 50 ml | 6x10-2M |
| Na2SiO | 122.06 g/mol | 0.05 g in 50 ml | 8.2x10-3 M |
| Citrate | 294.10 g/mol | 0.0147 g in 50 ml | 1x10-3 M |
I had the materials arrived today. Did an initial reaction today as a trial. I created the stock solutions using ddH2O and filling up the falcon tubes up to 50 ml. Then sonicated the calcium, phosphate, and metasilicate in the sonicator for 10 minutes. Mixed by vortexing.
- Added 4.06 ml of Igepal in 9.94 ml of cyclohexane to two 50 ml beakers.
- Began stirring, then added the calcium, phosphate, and metasilicate per protocol. No DNA origami was added. ALlowed to equilibrate for 30 minutes. The cyclohexane had evaporated significantly in this time.
- Mixed the microemulsions
- Quenched the reaction after two minutes with the citrate solution.
- Let the mixture continue for ten minutes with the citrate
- Ended microemulsion by adding 50 ml ethanol.
- Saw no particulates, so I transfered to a 50 ml centrifalcon tube(orange) and centrifuged for 10 minutes at 2,000 rpm. No pellet was formed. I do not believe I will be able to perform pellet purificaiton for the nanoparticles, since I believe that the origami and the nanoparticles will not pellet since they are so small.
Tuesday 6/11
Need to rethink the CPNP microemulsion protocol. Will start with just aqueous solutions.
- Trial 2 (50 ml falcon tube)
- Same as trial 1, used bulb pipette to mix the two microemulsions. Stored in a centrifalcon tube.
- Trial 3
- Made aq solution particles with no citrate present. Added 65 μL of the stock solutions to a 1.5 tube, and 1 ml of ddH20. Vortex. Will wait to see any visible particulate formation.
- Trial 4
- Same as trial 3 but added citrate after mixing reaction for 8 minutes. Should image.
- Trial 5
- Added 65 μL of the stock solutions, 50 μL of the origami non-purified after folding, 420 μL of ddH2O, and NO citrate was added.
- Trial 6
- To a 15 ml falcon tube with ~9ml of ddH2O, I added 100 μL of each stock solution (0.1M Calcium, 0.06M phosphate, 0.08 M silicate, plus 50 μL origami. After 2 minutes, 250 μL citrate was added.
Trial 5 showed significant aggolomeration of the DNA and calcium phosphate, so much so that it was cloudy and visible. It's somewhat nice to see, but I wonder if it's just the DNA agglomerating due to the high salt concentration or the calcium phosphate forming as well.
Trial 6 shows no visible particulates. Maybe it's working with nanoparticles, maybe it's too dilute.
Therefore, there is a shift in strategy: Focus on Aqueous reactions For tomorrow: Three groups of different stoichometry, three groups of DNA loading.
Wednesday 6/12
Will be meeting with Yun tomorrow at Nanotech West. Let's try to make a lot of samples. May want to include 1 ml of the microemulsion in cyclohexane. Let's try 994 ul of cyclohexane, 406 ul of igepal, and add 6.5 ul each of the calcium and 125 ul of the diluted phosphate/silicate.
Sample Group 1, aqueous: Created 9 groups of origami/CPNPs. Added DNA first to tube, then calcium followed by mix of phosphate and silicate for 1 ml total. Mixed for 1 minute at RT by vortex. Quenched with citrate for 1 minute. Sonicated for 15 minutes.
| ratios | 1.67:1 | 16.7:1 | 167:1 | quench 1 min |
|---|---|---|---|---|
| 0.5nM origami | 6.25 mM Ca, 3.75 mM PO4, 492 μM SiO3 | 6.25 mM Ca, 375 μM PO4, 49.2 μM SiO3 | 6.25 mM Ca, 37.5 μM PO4, 4.92 μM SiO3 | 225μM citrate |
| 0.5 nM origami | 625 μM Ca, 375 μM PO4, 49.2 μM SiO3 | 625 μM Ca, 37.5 μM PO4, 4.92 μM SiO3 | 625 μM Ca, 3.75 μM PO4, 0.492 μM SiO3 | 22.5μM citrate |
| 0.05 nM origami | 625 μM Ca, 375 μM PO4, 49.2 μM SiO3 | 625 μM Ca, 37.5 μM PO4, 4.92 μM SiO3 | 625 μM Ca, 3.75 μM PO4, 0.492 μM SiO3 | 22.5μM citrate |
Ran samples of these down the ethidium bromide gel. See lanes below.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ladder | 7249 pDNA | 0.5nM DNA/6.25nM Ca control | 0.5nM DNA/3.74 nM PO4 control | 0.5nM DNA/6.25nM Ca/10:6 | 0.5nM DNA/6.25nM Ca/100:6 | 0.5nM DNA/6.25nM Ca/1000:6 | 0.5nM DNA/625μM Ca/10:6 | 0.5nM DNA/625μM Ca/100:6 | 0.5nM DNA/625μM Ca/1000:6 | 0.05nM DNA/625μM Ca/10:6 | 0.05nM DNA/625μM Ca/100:6 | 0.05nM DNA/625μM Ca/1000:6 |

As one can see, lanes 14-15 of the 0.05 nM DNA are not visible due to the too dilute DNA. Lanes 7-9 seems to have the least free DNA, and lanes 11-13 have more visible bands and thus more free DNA. I suppose I'll take Lanes 7 and 9 on carbon film and see if I can see them on TEM.
It doesn't seem like this was a good assay. Hopefully DLS/Zetasizer will be more enlightening tomorrow on how the particles are.
Thursday 6/13
Will be going to zetasizer/dynamic light scattering training with Yun Wu today at 4:30pm. For the zetasizer I will be bringing specifically be a sample of a microemulsion that was in a 15 ml falcon tube: 3 ml of Igepal, 7 ml of cyclohexane, with 395 ul of water, 23 ul each of stock calcium, phosphate, and silicon, amounting to 464 ul of aqueous solution total. 80 ul of citrate was then added and the falcon tube was revolved for around 1.5 hours. This will be to measure the stability of the microemulsion.
For the dynamic light scatterer I will be measuring the tubes that were done yesterday at varying calcium/phosphate ratios and DNA loading. I will have sonicated these tubes again for 10 minutes prior to measurement, this will be the third time these particles were sonicated.
Finally, I will bring tubes of calcium phosphate nanoparticles that were synthesized as 6.25 mM calcium and 3.74 mM phosphate with and without citrate. This will be to see if we can measure a change in the zeta potential of the nanoparticles to confirm citrate binding.
Edit: Never mind, the training was rescheduled for Tuesday next week.
Friday 6/14
Started a 2.5 day folding for more bulk DNA. For one of the PCR strips I accidentally added 20ul of scaffolding to the first two tubes, and the 6th and 8th tubes have a little less scaffold as a result. These tubes were marked. Otherwise, the folding should be finished on Monday.
Here's a recent video from Adair's Keystone speech this year: [12]
Monday 6/17
- Purified bulk DNA from last week by PEG centrifugation. Pooled into 4 groups of 200 ul volume for the origami, added 200 ul of 15% PEG, and centrifuged for 30 minutes at 14.7k RPM. I then resuspended the pelleted origami/PEG in 200 ul of 1x FOB and 10 mM MgCl2.
- Will be trying to schedule AFM as well at nanotech west. Will need to think of ways to prepare the nanoparticles. Here's a good pdf on sample prep and AFM[13]
- Also prepared the stock solutions of calcium and phosphate again, they had a pH of around 10, which is somewhat surprising on how basic the solutions are by themselves. I hope this isn't affecting the DNA, but at least we definitely know now that the solutions are basic enough for CPNP synthesis. The phosphate and metasilicate solutions were combined together into one stock solution.
Tuesday 6/18
- Degassed water for one hour in the sonicator.
- Trial #1 - did microemulsion 10 ml in volume. used new solutions of calcium/phosphate/silicate. Reacted for 2 minutes. Citrate for 10 minutes. 4 ml of ethanol added. centrifuged 8,000 rpm for 30 minutes. Washed with ethanol twice. Redispersed in 1 ml of water. The pellet looked suspiciously gel-like, which is not the sort of pellet I've gotten from aqueous reactions only. I suspect it's mostly Igepal precipitate, but the pellet was rather small as well. I'll run a second trial, and if it does the same thing then I'll try using 50 ml falcon tubes, so that I can add signicantly more ethanol.
- Trial #2 - did microemulsion 10 ml in volume. new solutions were used plus 25 ul of the PEG purified bulk DNA was added. I equilibrated the phosphate/silicate/DNA for 30 minutes by rotary machine then added the calcium. reacted for 2 minutes, and then added the citrate. Quenched with 4 ml of ethanol. Continued for 10 minutes. Then centrifuged 8,000 as before. Problem: When I washed with ethanol, the pellet washed away with it. There was significantly more precipitate this time, and they definitely looked like Igepal precipitate to me. Which is perturbing...
- Trial#3 - did another microemulsion, but did not quench it with alcohol. I got good precipitate from it! I washed it three times with hexane, and then dried. It resuspended well in water too, with cloudiness at 500 ul. No DNA origami involved.
- Trial #4 - I replicated trial #3. Ran reaction for around 2 minutes, citrate 10 minutes, 8,000 rpm for 30 minutes. Hexane washed pellet. Dried for 10 minutes. Resuspended in 1 ml. This was a good concentration, not as cloudy as trial #3. GREAT!
- Finally, I performed four 1 ml aqueous reactions of CPNP with and without DNA origami, and with or without citrate dispersion. Mixtures were 712 ul of ddH2O, 62.5 ul calcium, 125 ul of PO4/SiO3, and 225 ul of citrate. They were reacted for around 2 minutes, and citrate was then added.
Here are the zetasizer results. I'll upload the DLS data tomorrow.
Trial #3 No origami, CPNP by microemulsion. Zetasizer after 5 rounds.
| Mobility | Zeta Potential(mV) | Rel Residual | |
|---|---|---|---|
| Mean | (-1.51) | -21.23 | 0.0185 |
| Std Error | 0.07 | 0.92 | 0.0013 |
| Combined | (-1.51) | -21.18 | 0.0104 |
Trial #4 +origami, CPNP by microemulsion. Zetasizer after 5 rounds.
| Mobility | Zeta Potential(mV) | Rel Residual | |
|---|---|---|---|
| Mean | (-2.56) | -36.00 | 0.0212 |
| Std Error | 0.15 | 2.09 | 0.0036 |
| Combined | (-2.56) | -35.98 | 0.0129 |
Aqueous CPNP, no microemulsion, +DNA but no citrate. Zetasizer after 5 rounds.
| Mobility | Zeta Potential(mV) | Rel Residual | |
|---|---|---|---|
| Mean | (-0.77) | -10.86 | 0.0168 |
| Std Error | 0.09 | 01.29 | 0.0027 |
| Combined | (-0.75) | -10.52 | 0.0111 |
^ This is good, what we should be seeing. Without citrate, the CPNPs should have a more positive zeta potential. The microemulsion CPNPs also demonsrate a negative zeta potential that is less than -30 mV, which is a good sign for dispersion! I now need to tighten up the protocol to gain more monodispersed particles, and I also need to effectively demonstrate the zeta potential changing with and without citrate!
Here's a good paper on how to measure nanoparticles by AFM: [14]
Wednesday June 19th
Apparently, the blue polystyrene falcon tubes are not compatible with my microemulsions! I added cyclohexane, Igepal, and water...but the microemulsion was cloudy throughout the reaction with very bad precipitation after centrifugation. I've been using the more expensive polyethylene centrifuge falcon tubes in the past, which have always had great clear micreomulsions.
Ran four different CPNP microemulsion groups today. With and without citrate, with and without DNA origami. The emulsions are a little bigger now, I'm using a falcon tube each for microemulsion A and B, and then mixing them together in a 50 ml falcon tube. So the reactions are about twice as large now, but now I'm adding equilibrated microemulsions A and B together, rather than adding the calcium or phopshate directly into the microemulsion. Hopefully this will lead to better equilibrated reaction conditions during the actual mixing, and I'll see better monodispersed particles.
Zeta Potentials: (I may have mixed the +citrate/origami DLS sample with the +citrate DLS sample. I've switched their results for the time being.)
| Mobility | Zeta Potential(mV) | Rel Residual | |
|---|---|---|---|
| Mean | (-1.36) | -19.11 | 0.0168 |
| Std Error | 0.04 | 0.60 | 0.0017 |
| Combined | (-1.36) | -19.05 | 0.0127 |
| Mobility | Zeta Potential(mV) | Rel Residual | |
|---|---|---|---|
| Mean | (-1.60) | -22.53 | 0.0174 |
| Std Error | 0.07 | 1.00 | 0.0031 |
| Combined | (-1.60) | -22.49 | 0.0092 |
| Mobility | Zeta Potential(mV) | Rel Residual | |
|---|---|---|---|
| Mean | (-1.71) | -24.02 | .0.0194 |
| Std Error | 0.03 | 0.44 | 0.0023 |
| Combined | (1.71) | -24.00 | 0.0129 |
| Mobility | Zeta Potential(mV) | Rel Residual | |
|---|---|---|---|
| Mean | (-2.05) | -28.74 | 0.0167 |
| Std Error | 0.09 | 1.28 | 0.021 |
| Combined | (-2.04) | -28.72 | 0.0113 |
The nanoparticles by the DLS were still too polydispersed, but we're getting close I think. I'm getting a polydispersity of ~0.3, where <0.1 is the expected value for mondispersed nanoparticles. Tomorrow, I will make a more concentrated solution of citrate so that we don't have to add so much more additional aqueous solutios, thereby keeping the microemulsions smaller. I'm worried the citrate groups may have bigger nanoparticles simply the addition of more aqueous environment. I will also lower the centrifuge time to 15 minutes, and add the sonication method. The collected nanoparticles will be dispered in a larger volume of two 1.5 ml tubes.
If they're still too polydispersed, it may simply just be a relic of the pelleting by centrifuge. I may also want to try once again to quench the reaction by ethanol by centrifuging.
Thursday 6/20
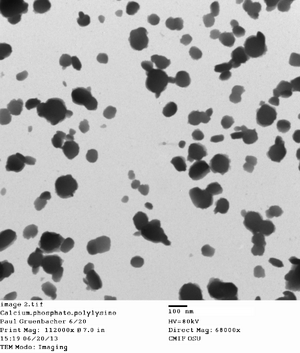
Ran a CPNP microemulsion. Made a solution of 0.01 M citrate, thus only adding 16 ul of it to the reaction mix. I sonicated it for 30 minutes, which seemed to make optically clear. The DLS suggested small particle sizes, but that may be due to some dust that I noticed in the tube. I also took TEM images, but I'm worried there may be UFo crystallisation.
 |
 |
 |
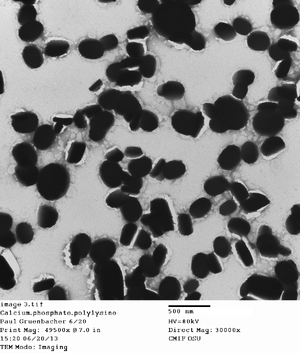 |
This appears to be UFo crystallization...but they are around the same sizes I would have expected from the particles
Here's some promising DLS results for one of the sonicated CPNPs. A polydispersity of 0.193 is a good move towarods a monodispersed CPNP of <0.193


Monday 6/24
I ran another CPNP with DNA origami, sonicated for 30 minutes. However the DLS results were bad, and so I think that settles the issue that ultracentrifugation is not appropriate for purifying the nanoparticles. Perhaps if they had the PEG ligands prior to the centrifugation, then they would be properly dispersed after. However, with charged particles during centrifugation, there is significant flocculation to be expected, and so I need to either explore ligand attachment while still in the microemulsion or by column method. Will being searching for commercial products.
Here is the file from the first three sessions with the DLS from 6/18-6/20: File:20130618.doc
Here is the file from 6/24 today DLS: File:6-24.doc
Tuesday 6/25
Ran TEM of CPNP. Incubated grid on 1% polylysine droplet for 4 minutes, washed with water three times, and then placed 10 ul of CPNP on it. NOTHING on the TEM grid visible other than what appears to be a layer of polylysine. I don't know if the CPNP aliquot was just too dilute, but I expected some particles to have adhered... very frustrating.
Wednesday 6/25
I have AFM training at 2pm today. Meeting with Bryant of Dr. R Lee's lab at 11am tomorrow
Purchasing information for Bio-Rad econo column:
| Item | Catalog | Price | Notes | link |
|---|---|---|---|---|
| 1.5 x 10 cm glass econo-column | 737-1512 | $77 | pkg of 2, includes stopcocks, 18ml | [15] |
| 1.5 id Flow Adaptor | 738-0016 | $125 | 1-14 cm length. see video: [16] | [17] |
| Cole-Parmer tubes and fittings | EW-45502-04 | $8.00 | female luer x 1/8" barb, nylon | [18] |
| Cole-Parmer tubes and fittings | EW-45505-56 | $5.00 | Male luer lock plug | [19] |
| Cole-Parmer tubes and fittings | EW-45502-28 | $7.25 | Female luer cap, nylon | [20] |
| Cole-Parmer tubes and fittings | EW-06605-29 | $61.00 | 25ft/pkg, 1/8" x 3/16" | [21] |
| Polyscience Glass beads | 18900-100 | $104.00 | 210-250 μm beads, 100 g | [22] |
US. Plastic Corp. 1/4" polypropylene tubing [23] Will also need clamps, ring stand, funnel(s), rubber stoppers, and ETHANOL.
I will be using these with the syringe pump and 10-50 ml disposable syringes. If gravity is adequate for the flow rate, then we'll need to use a funnel or reservoir, but I see no point in spending $190 on a funnel when one can be found around the lab. Fraction collection will have to be manual.
I may also want to treat the glass spheres with APS in glacial acetic acid, and ddH2O. Can then be oven-dried at 70°C. According to Xia Cao [24].
Also started working on AFM protcol OhioMod2013:Methods/AFM.
Thursday 6/27
There is an Agilent 720-ES ICP-OES (Inductively coupled plasma, optical emission spectroscopy) for which I can use to determine the concentration of the calcium and phosphate in the sample.
Fluorescent Tracing
- Fluorescein 494 nm -> 521 nm, ~$24 for 100 grams. Adair, Bioconjugation
- Indocyanine Green. Infrared. Adair, Bioconjugation.
- Cy3 amidite loaded in CPNP. Adair, Fluorescent markers (2008).
- Propidium Iodide 535 nm ->617 nm.
I put in the order for the columns.
Got liposomes from Bryant Yung of Dr. Lee's lab. Two tubes of:
- QTCN: Quaternary-Tertiary Lipid Amine Cationic Nanoparticles (1 mg/mL)
- SPLN: Small Peptide Lipid Nanoparticles (2 mg/mL)
both are in 40% ethanol in 10 mM citric acid buffer (pH of 5). I believe the lipid mixture is composed of DODAP, Lac-DOPE, DOPE, DMG-PEG, and with or without gramacidin A, at a molar ratio of 50:10:28:2:10 respectively.
- Take lipid stock, combine with oligo at ratio of 1:15 DNA/lipid(weight/weight) in 10x transfection medium.
- Set aside 10-15 min to allow for electrostatic interaction.
- Dilute to final volume
- Extrude through polycarbonate filter, or other sterile filter in general
They can then be lyophilized with 10% sucrose as the lyoprotectant for long-term storage.
Friday 6/28
Submitted new design for overhang staples for the AlexaFluor555 staple extension: File:66 helice final v6.json. Staples were ordered by Carlos. Here is another file with some extra overhang before they were trimmed:File:66 helice final v5.json Here is the excel file used for ordering: File:CalcPhos origamiv3 orderform 6-28.xls Were ordered from Operon.com at 10 nM, HSPF purified, wet conditions, at 100 μmol/L.
Info on the Alexa: C - OrangeBox D1_21_5 Alexa555 GGGATTTTAGACAGGAACGGT ordered:11-8-11
Can always buy a fraction collector significantly cheaper off of Ebay or used depot stores. [25]
Tuesday 7/2
Started working on the liposomes. Here's a protocol: OhioMod2013:Methods/Liposomes
Can buy the round-bottom tubes and other basic stuff at the Chem Store in Celeste Lab
Wednesday 7/3
Learned scaffold prep protocol
Attempted the encapsulation liposome efficiency trials, but it is evident the liposomes are not forming or not protecting the DNA from the YOYO marker.
Here are two files that I've made from some that appear to be working, but overall there is some sort of difficulty with this assay. trial 9 and trial 8
I had talked to Bryant Yung about the issue, and he has made useful suggestions.
I now resuspended the DNA structures in 10 mM citrate buffer at 40% ethanol concentration with a pH of ~4. The lipid/DNA mixture was then diluted with 10 mM citrate buffer pH ~4 with no ethanol. This seemed to protect the DNA better, but I am still having difficulty demonstrating an increase in flourescence after addition of the detergent Igepal
Friday 7/5
Important note: There can no longer be any EDTA present when resuspending the DNA! The EDTA will chelate the calcium ions! I just realized this unfortunately, and since the EDTA is at 1 mM concentration it can have a large affect. I will now only be PEG purification of the DNA origami, and re-suspending in tris buffer without EDTA.
Ran new strip of DNA origami.
Monday 7/8
- Created 10 mM tris-HCl buffer with 10 mM Mg Cl2 at ph 7.85
- Added 10 mM MgCl2 to the 10 mM citrate buffer with 40% ethanol (realized that my structures are probably easily dissolving when being resuspended without the MgCl2)
- Added 10 mM MgCl2 to the 10 mM citrate buffer.
- Fulfilled oligo-us order form for the new origami overhang staples. Saved email and order forms.
- Finally some decent results for the liposomal encapuslation efficiency.
- Basically added 2mg/ml lipids at varying volumes to 4 ul of DNA origami that had been resuspended in 10 mM citrate with 40% ethanol and 10 mM MgCl2. Detergent was then added to the second portion of each group in step 2, including the second part of the positive 2 ul origami group and 4 ul origami group. Funnily, the YOYO flourescence seems to decrease with ever increasing staple size, confirming some suspicions earlier. It may be too much origami or DNA present may start to lower the YOYO flourescence. There must be some peak concentration of DNA.
- Excel file [26] on google docs.
- Also, did scaffold prep today for 7560

Tuesday 7/9
Started as 2.5 day folding with just core staples of 56 helice structure for Danny's polymerization. We'll do the polymerization next week on Friday.
Here's the DLS of the liposomes. 10 ug of lipids were added to 15 ul of DNA origami. Raised to 1 ml of the 10 mM citrate buffer at low pH of 4.25
| Liposomes with origami | Liposomes without origami |
|---|---|
| 4.35 mV zeta +- 1.23 | 8.72+-0.72 mV zeta |

|

|
Wednesdday 7/10
Decided to start the column without the clamp stand, just built one out of cardboard that works just fine.
- Fill column with ethanol. Then add 10 grams of glass microspheres directly into the column, no need to worry about making a slurry. Elute the column somewhat to let glass settle but leave it filled with ethanol.
- Add the flow-adaptor, slowly add it into the ethanol so that there are no air bubbles and so there is ethanol exiting from the tube on top of the adaptor.
- Close flow-adaptor on top of the glass stationary bed.
- I then created all new solutions of calcium, phosphate, and citrate.
- Created CPNP micromeulsion, equilibrated A and B for 30 minutes, mixed for 2 minutes, and added 16 ul of 0.01 M citrate for 10 minutes. Then added 40 ml of ethanol to quench.
- Picked up solution into 60 ml syringe.
- Inserted into column at slow flow rate, collecting in the first 15 fractions tubes. Collected those into 1.5 ml tubes, and then dumped the fraction tubes and washed them.
- Washed the bed with ~20 ml of ethanol
- Eluted the column with ~40 ml of 70:30 ethanol-water
The third fraction during the 70:30 elute had a very high DLS concentration. Suprisingly high concentration, and it became cloudy after the addition of water while in the cuvette. However the zeta is confirmatory that it is negative particles, and so we have a good indiciaiton that the column is working.
Restuls: -29.28+-1.47 mV zeta potential for the particles.


Thursday 7/11
Did another column run, this time without any air bubbles. I did this by raising the flow adaptor so that there is an air pocket followed by an aqueous layer over the stationary bed. This will allow space for air pockets to enter without entering the glass beads, while still minimizing the amount of free fluid above the stationary. It seems to work very well. Once again I got CPNP collection in the third tube fraction. Here are the DLS results. Now I just need to figure out how to change the viscosity and refractive index of the DLS to create better results, but at the moment the software won't allow me to do so and I need Yun's help. Also, I need to determine how to change the solution suspension if I so desire.


Monday 7/15
- Performed CPNP purification. Got new clamp stand today woohoo!
- Performed overhang folding reaction 2.5 days 14 magnesium. 16 overhange staple tubes are stored on shelf in fridge. Made 5 new prestocks of core from p085, core from p086, neighbor, polymer, and overhang staples.
- Performed polymerization 40°C ramp overnight for Danny. Created a new pre-stock of the neighbor staples in well A5, and of the polymer staples in A6, added them at estimated 10x fold to the gel-purified core structure that had been folded last week. The gel image looks good, here it is:
.jpg)


DLS useful guide [27]
Wednesday 7/18
Add 7.2 ul of 5 uM YOYO for every 50 ul of 1 nM origami for 1:10 dye to base efficiency.
Friday
Finished folding the overhang structure. Ran gel at 70 volts for 3 hours to make sure the structure folded. I'm going to do a quick PEG purification on one of the tubes and incubate it with the Alexa staples so I can do a quick test today to see if it works. Will use flourescent spectrometer at nanotech west, to see if the flourescence is in the origami as well as when run with the CPNP reaction. Felix software manual for flourescence: [28] and the link to the Timemaster spectroflouremeter [29]
- Later: Apologies, I've been writing less on the notes.
I've been adding incorrectly too much phosphate and silicate. Should instead be 46 ul instead of 92 ul, I had made a very simple mistake of not understanding solutions. The ratio should be back to 10:6 now and at 9.4 mM, 5.6 mM, and 0.77 mM for calcium, phosphate, and silicate respectively. I do no think the extra phosphate would have caused any significant problems and I expect similar nanoparticle results for next week, but will be interesting to see if there are major changes.
I think the Alexa 555/565 peaks are somewhat problematic on hose close they are, I had zero 565 peak visible even in the positive control solution for the fluoremeter at nanotech west. I may need to move the laser excitation farther away to 545 perhaps.
I'm also not sure whether to be adding DNA to the calcium or phosphate solutions during equilibration, there has been no agglomeration evidence for either microemulsion, and I've had normal nanoparticles for both. I need to add the DNA origami to a calcium solution and check to make sure they're not being torn apart by the calcium
Monday 7/22
So it seems that I shouldn't be PEG purifications, and should just stick with the gel purification method based on this gel I ran.
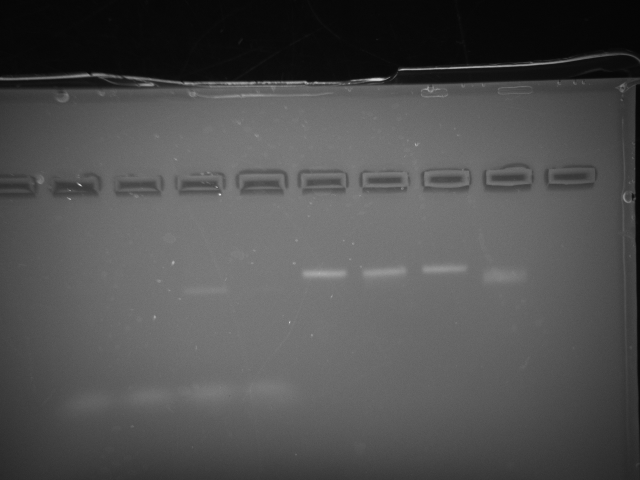
Lane 2-5, PEG purified structures. Lane 6-9 Gel purified. They are in order of positive control at 5 ul origami or (~5 nM origami), 5 nM origami to 10 nM calcium solution, 5 nM of origami to 6 nM phosphate, and 5 nM origami to 10 mM citrate at pH of 4.25. Of the gel-purified structures we see tight bands for the first three, with only degradation to the citric acid exposed DNA. The magnesium concentration of the calcium and phosphate groups would have been ~2.33 mM which may have helped keep the tight bands as well but at least we know the calcium and phosphate aren't tearing them up. A later time period should probably show increased degradation of the citric acid exposed DNA, which is probably disocciating it's hydrogen bonds due to increased protonation. This is problematic for the liposomal strategy of delivering the origami. For the PEG purified structures we see the staples but poor bands.
I'm running another group of 5 ul 1 nM origami in a 1ml 30% microemulsion to test if the Igepal will deissociate the origami. I mixed the microemulsion for around 5 minutes, then took 30 ul aliqouts, added 120 ul of FOB to break up the emulsion. Then spun to seperate the aqueous phase from the now coagulated emulsion. It's possible the origami was stuck in the coagulant, we'll see. I then took the ~100ul aqueous phase and added 10 ul glycerol to it. I then loaded in gel lanes, ran for a few minutes, and then added more of the samples to the lanes. This is to compensate for the multiple dilutions of the origami, which would now be like 1.25 pM for every load I did. Yikes, I probably won't see anything.
Here are the overhang structure forms: [[30]] and [[31]]
L James Lee, nsecpti:. 3 power switches ignite button first. should read 70 watts. set shutter to open/closed when loading and reading. Turn on PTI Felix software admin pass: pti. Acquisition-> new acquisition. Set to emission scan, digital YW, integrate 05 sec, one step turn. seperate excitation and emission by around 50 nm.
The Alexa 5555 i am using is from the C-orange box D1_21_5' Alexa_555. Made a 100-dilution of hte 100 uM solution to a 1 uM aliquot.
Tuesday 7/23
There is also a UV-vis spectrophotometer at Nanotech West. When I read the nanoparticles I had a 1.165 A260 and 2.2224 A280, which makes sense since the larger UV will pick up on the nanoparticles. Perhaps if I set a baseline the DNA might be detected that way.
Made new folding reaction of the overhang structure.
Made new CPNP wit h15 ul of core gel-purified origami loaded in calcium microemulsion. Used new glass beads for this to see if there is an improvement, in which case I should start washing the other beads more thoroughly.
Will dissolve the CPNPs using a 100 mM EDTA solution or 10 mM citric pH 4.25 buffer solution.
Made EDTA solution. 1.872 grams in 50 ml of ddH2O. Raised pH to 6.10.
Here's a document on the CPNPs I did today, very good results with low polydispersity, and the zeta potential was effective. From now on I'll be assuming a dielectric constant of 31 for 70% ethanol.
I also got a 1 ml dialysis bag for which to hopefully dialize the solvent of 70% ethanol in ddH2O. The surrounding water had 1.3 mM phosphate and 2.2 mM calcium to hopefully keep the osmolality balanced. The nanoparticles themselves shouldn't necessarily affect the osmolality. I'll probably run the dialysis for overnight but I'll check after a couple hours to see if the sausage increases in size, which would be a bad sign.
Later: There was some definite increase in the size of the dialysis tubing demonstrating osmolality differences. However it seems to have worked well as there were no cloudiness or emulsions formed when addding the aqueous citric buffer and EDTA.
Wendesday 7/24
Created a 34 ul aliquot of the DNA overhang structures plus 1 ul of the Alexa 555 oligos for a 30 fold annealing. They should anneal at room temperature.
There is some flourescence of the Alexa 555 on the RT-PCR actually, about a 1000 increase in RFU.
Will go to measure the DLS again today after 24 hours and see if the nanoparticles are fullly dissolved with the citric acid + EDTA
Thursday 7/25
Set a new CPNP synthesis today with 15 ul of Alexa 555 overhang structures. They only had about 10 minutes to anneal before the reaction started, so hopefully that was enough. Even if they don't anneal with the structure, they should still be taken up by the particles and fluoresce when I centrifuge the particles after the column. I'll resuspend the particles to make them more concentrated so the Alexa will hopefully show up on the RT-PCR.
Hmm, no pellets were formed after centrifuging. I suppose the nanoparticles by themselves will not be able to be ultracentrifuged.
Got some more dialysis tubing from Poirier's lab, it seems to work well. Here is the link to the tubing: [32] and the clips [33]. I may just try to reuse the three that I have though since they're rather expensive.
I'm going to make a 1% agarose gel. Make some custom lanes to see if I can dissolve the calcium phosphate and then separate the DNA to measure it, assuming it's still been encapsulated. I'm having my self-doubts now.
Hilarious calcium video: [[34]] and my all-time new music station: [[35]]
Thursday 8/1
Gel-purified core and overhang structures.
Made 20 liters of TAE buffer with and without magnesium each.
Created CPNP without origami, will dry on coverslip for AFM training today.
Friday 8/2
Made 1 ml and 400 ul aliquots of the 1.375 M MgCl2 for gels.
Performed Dialysis of the CPNPs for 3 hours.
Thursday 8/9
So loading the CPNPs with daunorubicin worked well on the PTI fluorometer, with distinct flourescent peaks observed. Unfortunately this was also seen in CPNPs with daunorubicin loaded but without DNA origami present, so the daunorubicin at high concentrations is being loaded into the CPNP quite regularly. HOwever, it does lead to interesting flourescent nanoparticles theoretically.
I also tried testing Alexa 555 overhang structures on the fluoremeter, and I got a much wider and less distinct fluorescent peak. I can't tell if this is perhaps aqueous artificial flourescence, so I'm making CPNPs today with no Alexa 555 in them to serve as a negative flourescent control. I'm also making another Alexa 555 loaded CPNP group, with incubation of the Alexa 555 and overhang structure for more than an hour, which I think is suffifcient for binding to the origami. Hopefully we see something distinct today to declare origami in the CPNPs. Added 6 ul of 1 uM Alexa 555 to 50 ul of overhang structure. I think yesterday it was 5 ul of 1 uM alexa to 40 ul of overhang structure, so hopefully we see higher peaks.
Also, when performing DLS on the CPNPs after column purification, there is not much kilo-counts per second, but after dialysis for 30 minutes it goes up and is cloudy. The next day it is less cloudy with normal kilocounts, and further dialysis for >6 hours leads to the solution being able to run in a gel. Needs 6 hours of dialysis at least then..
Monday 8/12
Gel purified 6 hb, 12 hb, and 18 hb structures for OhioMod.
Did gel-purified overhang structures in 10 mM calcium, 6 mM phosphate, and in 8.2 mM citrate to replicate previous results. The magnesium from the gel purification is still present, so seeing tight bands while good is certainly not conclusive. update: good bands!
Also did 6 hour dialysis on CPNP with Alexa 555, will filter after and perform DLS, Zeta, and flouremeter this evening to check. Update: Well nevermind, it was just cloudy. May try to dialyze again?
Wednesday
Performed Danny's project follow up, added 20 ul of core 1 nM and 1 ul of 500 nM neighbor and polymers at room temperature for 3 hours 11:30 to 2:30, then performed negative TEM stain for them.
Tried to PEG-purify overhang structures for 40% ethanol citric resuspension. Then did 4 ul of 1 nM origami in 40% ethanol suspension with 0.5 ul of 2 mg/ml lipids, 15 minutes, re-suspend up to 50 ul of citric buffer. Will do TEM stain as well with the polymer strands at 2:30
Thursday
Did liposome suspension again today at 10am, then did detergent assay.
Began folding new overhang structures for over the weekend at 16 mM MgCl2
Took 80 ul of overhang structure, added 16 ul of 10 mM daunorubicin. THen added to CPNP mixture to check on fluorometer (will check to make sure signal exists before making the control to conserve time). Used control CPNP with just the 16t ul daunorubicin added as well.
References
- Dworetzky SI, Lanford RE, and Feldherr CM. The effects of variations in the number and sequence of targeting signals on nuclear uptake. J Cell Biol. 1988 Oct;107(4):1279-87. DOI:10.1083/jcb.107.4.1279 |
-
pmid=PMC65638
- van Gaal EV, Oosting RS, van Eijk R, Bakowska M, Feyen D, Kok RJ, Hennink WE, Crommelin DJ, and Mastrobattista E. DNA nuclear targeting sequences for non-viral gene delivery. Pharm Res. 2011 Jul;28(7):1707-22. DOI:10.1007/s11095-011-0407-8 |
- Ritter W, Plank C, Lausier J, Rudolph C, Zink D, Reinhardt D, and Rosenecker J. A novel transfecting peptide comprising a tetrameric nuclear localization sequence. J Mol Med (Berl). 2003 Nov;81(11):708-17. DOI:10.1007/s00109-003-0483-2 |
- Sebestyén MG, Ludtke JJ, Bassik MC, Zhang G, Budker V, Lukhtanov EA, Hagstrom JE, and Wolff JA. DNA vector chemistry: the covalent attachment of signal peptides to plasmid DNA. Nat Biotechnol. 1998 Jan;16(1):80-5. DOI:10.1038/nbt0198-80 |
- Chen X, Kube DM, Cooper MJ, and Davis PB. Cell surface nucleolin serves as receptor for DNA nanoparticles composed of pegylated polylysine and DNA. Mol Ther. 2008 Feb;16(2):333-42. DOI:10.1038/sj.mt.6300365 |
- Liu G, Li D, Pasumarthy MK, Kowalczyk TH, Gedeon CR, Hyatt SL, Payne JM, Miller TJ, Brunovskis P, Fink TL, Muhammad O, Moen RC, Hanson RW, and Cooper MJ. Nanoparticles of compacted DNA transfect postmitotic cells. J Biol Chem. 2003 Aug 29;278(35):32578-86. DOI:10.1074/jbc.M305776200 |
- van Steenis JH, van Maarseveen EM, Verbaan FJ, Verrijk R, Crommelin DJ, Storm G, and Hennink WE. Preparation and characterization of folate-targeted pEG-coated pDMAEMA-based polyplexes. J Control Release. 2003 Feb 21;87(1-3):167-76. DOI:10.1016/s0168-3659(02)00361-9 |
- Motskin M, Wright DM, Muller K, Kyle N, Gard TG, Porter AE, and Skepper JN. Hydroxyapatite nano and microparticles: correlation of particle properties with cytotoxicity and biostability. Biomaterials. 2009 Jul;30(19):3307-17. DOI:10.1016/j.biomaterials.2009.02.044 |
- Chan CK, Senden T, and Jans DA. Supramolecular structure and nuclear targeting efficiency determine the enhancement of transfection by modified polylysines. Gene Ther. 2000 Oct;7(19):1690-7. DOI:10.1038/sj.gt.3301275 |
- Bisht S, Bhakta G, Mitra S, and Maitra A. pDNA loaded calcium phosphate nanoparticles: highly efficient non-viral vector for gene delivery. Int J Pharm. 2005 Jan 6;288(1):157-68. DOI:10.1016/j.ijpharm.2004.07.035 |

{kind=link}