Isaiah M. Castaneda Week 9
From OpenWetWare
Jump to navigationJump to search
Media:HIV_Structure2011_CRIC.ppt
- Convert your DNA sequences into protein sequences. How will you do this? How will you know that it was done correctly?
- The protein sequences that we need can be found off the BEDROCK site. The BEDROCK project is funded by the NSF, so I would hope that the information they provide is reliable. The sequence data is apparently very commonly used, so it is likely widely accepted as correct. If access to this data was not available, there could have been room for error. Some sort of program would need to be used to translate the nucleotide sequences into the corresponding amino acid sequence. Some unaware, yet dedicated people may even venture so far as to derive the amino acid sequence themselves. Getting the information from a reputable source is a much safer way to go about it.
- Perform a multiple sequence alignment on the protein sequences. Are there more or fewer differences between the sequences when you look at the DNA sequences versus the protein sequences? How do you account for this?
- There are fewer differences between the protein sequences compared to the DNA sequence differences. This is because there is the fascinating characteristic known as redundancy in the amino acid code. This describes instances when more than 1 codon codes for the same amino acid. So, although the nucleotide sequence changes, it doesn’t always alter the amino acid it codes for.
- Which of the procedures from Chapter 6 that you ran on the entire gp120 sequence are applicable to the V3 fragment you are working with now? How are they applicable?
- The ProtParam could provide some useful information about the V3 region. One example is the instability index. It would certainly be of interest to know the stability status on the V3 regions being looked at and if certain protein sequence changes have a correlation with protein stability. Also, knowing if there are Transmembrane regions could provide further information on protein function.
- Chapter 11 contains procedures to use for working with protein 3D structures. Find the section on "Predicting the Secondary Structure of a Protein Sequence" and perform this on both the entire gp120 sequence and on the V3 fragment that we are now working with. You will compare the predictions with the actual structures.
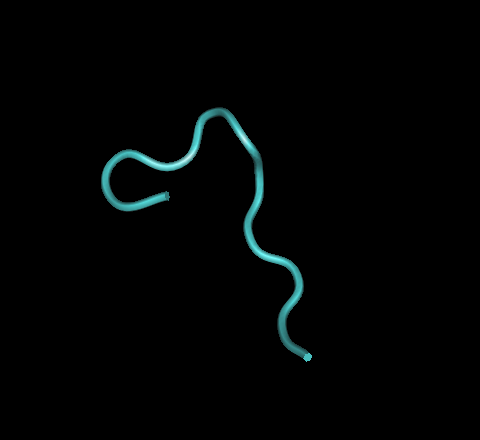
- I pointed my browser to bioinf.cs.ucl.ac.uk/psipred/. I then pasted the 1GC1 sequence into the sequence window. The 1GC1 sequence is from the Kwong paper and contains the gp120 protein. After hitting “predict,” I received an error message. I had foolishly forgotten to enter an identifier. After correcting the issue, I re-submitted the information. About 10 minutes later, I received my results. A .PDF of the results may be viewed HERE. I compared some of the predicted features with actual information from the following web page, http://www.ncbi.nlm.nih.gov/protein/3319094?report=graph#. What I found was that when confidence of prediction levels were high, the predicted structure was very similar to the actual structure, but when confidence levels were lower, the predicted structure was less likely to match the actual. For example, a helix was predicted around amino acids 116-119 with a low level of confidence. In the actual structure, they are not present. This tool appears to have a very decent level of accuracy. I repeated the same process with a V3 sequence from the Stanfield 1999 paper. The PDF file can be found be clicking HERE. The prediction shows that the protein should consist of only strands and coils & that is what the image of the actual structure shows.
- Download the structure files for the papers we read in journal club from the NCBI Structure Database.
- Kwong et al. (1998) structure 1GC1.
- Stanfield et al. (1999) structure 1F58
- Stanfield et al. (1999) structure 2F58
- Stanfield et al. (2003) structure 1NAK
- All structure files have been downloaded. I followed each link and clicked the appropriate structures.
- These files can be opened with the Cn3D software site that is installed on the computers in the lab (this software is free, so you can download it and use it at home, too.) Familiarize yourself with the software features (rendering and coloring) with both the gp120 peptide and ternary complex structures. Alternately, you may choose to use the Star Biochem program to do this portion of your work. Answer the following:
- Find the N-terminus and C-terminus of each (poly)peptide structure.
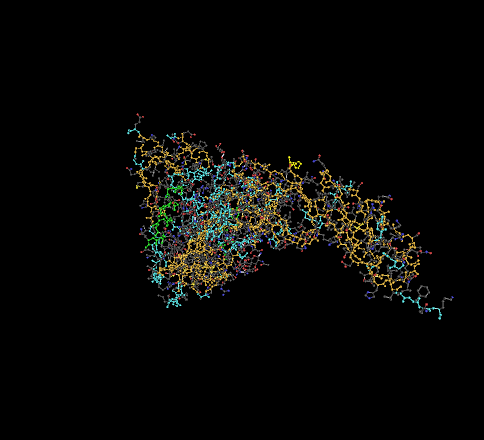
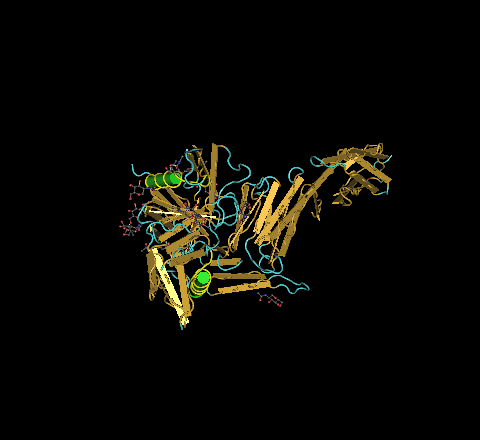
- 1GC1 - The N-terminus is located on the bottom right of the picture shown below. The C-terminus is located on the upper left.
- 1F58 - The C-terminus is the highlighted portion on the left, the N-terminus highlighted portion on the right.
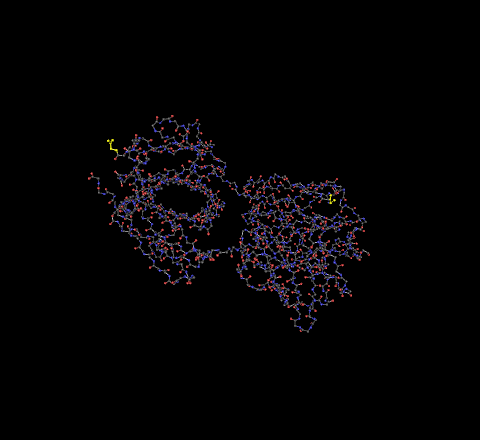
- 2F58 - The C-terminus is the highlighted portion on the left, the N-terminus highlighted portion on the right.

- 1NAK - The C-terminus is the highlighted portion on the left, the N-terminus highlighted portion on the right.
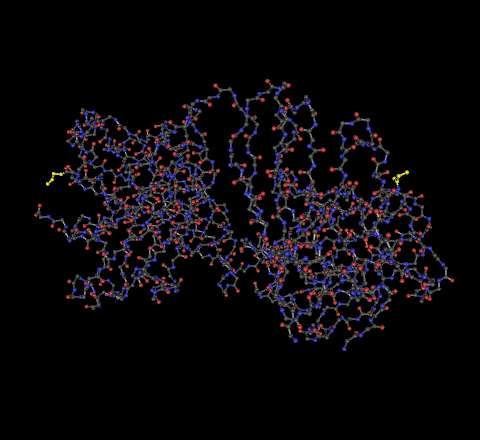
- Locate all the secondary structure elements. Do these match the predictions you made above?
- Using the predict protein predicting tool from chapter 11, the results, which may be seen found above in a .pdf link, showed that the gp120 protein consists of coils, helices, and strands. This proved to be true. To further show that the predictions had a sound level of accuracy, in the picture below, there are two highlighted structures. The prediction indicated that these sequences would be involved in helices. Clearly, this is what has occurred.
- The V3 regions from the Stanfield papers can be seen by clicking the following links
- They were found by specifically choosing the HIV-1 protein chain and deselecting the Fab chains.
- Once you have oriented yourself, analyze whether the amino acid changes that you see in the multiple sequence alignment would affect the 3D structure and explain why you think this.
- When working on the HIV Structure project, I performed 14 secondary structure predictions among various visit strains from subjects 7 & 13. They can be found HERE. What I found was that despite many amino acid changes, secondary structure was conserved. This is because single amino acid changes are not enough to create that big of a change in secondary structure. However, when amino acids change, the side chain types do as well. Since side-chains determine folding patterns, it is my belief that amino acid changes would result in 3D structure change, although it may not be drastic.
- The journal club papers we read are quite old already for a fast-moving field. Using the Web of Science (or PubMed or Structure) databases, find at least one more recent publication that has a structure of gp120 (V3) in it and download the structure file to view. What additional information has been learned from this new paper?
- The paper chosen is entitled, "Clinical resistance to vicriviroc through adaptive V3 loop mutations in HIV-1 subtype D gp120 that alter interactions with the N-terminus and ECL2 of CCR5," by Ogert et al., 2009.
- There are 6 amino acid changes within the V3 loop that are associated with a defective virus
- One of these changes is within the highly conserved GPGR region
- 3 of the observed changes are enough for partial immunity
- 4 of the observed changes result in nearly full immunity
- these changes affect the V3 region's ability to bind to CCR5
- The paper can be found here, http://0-www.sciencedirect.com.linus.lmu.edu/science/article/pii/S0042682210000863
- Below is the structure of the GP120 protein with V3 region from the subject of interest, subject 91. This subject was studied because there was nearly complete viral suppression
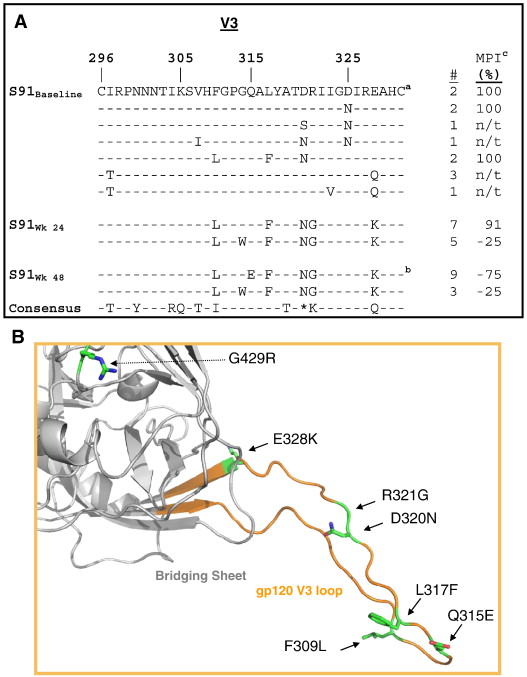
- The paper chosen is entitled, "Clinical resistance to vicriviroc through adaptive V3 loop mutations in HIV-1 subtype D gp120 that alter interactions with the N-terminus and ECL2 of CCR5," by Ogert et al., 2009.








{kind=link}
{kind=link}
{kind=link}