IGEM:MIT/2006/Index
From OpenWetWare
Jump to navigationJump to search
Protocols
- Smell Tests - André
- Concentrations
- Isoamyl Alcohol: 10mM desired final concentration; 4.4μL stock IA-OH per 10mL culture
- Salicylic Acid: 2mM desired final concentration; 40μL 0.5M SA or 200μL 0.1M SA per 10mL culture
- Concentrations
- PCR - Veena
- First, you might want to dilute some of your miniprep product (that is, your plasmid DNA...this is your template) to a concentration of 1ng/μL, and label this tube; for example, if I have BSMT plasmid DNA that I got from a miniprep and it's at a concentration of 50ng/μL, I'd label a new tube "BSMT for PCR" and add to that tube 49μL of water and 1μL of the 50ng/μL plasmid DNA.
- You should also prepare 25μM tubes of primer, if you don't have them already.
- In each tube, you want a total reaction volume of 50μL, with 1ng of your template DNA, .6μL of each primer (forward and reverse) (.6 μL of the 25μM primer yields roughly 300 nM final concentration of that primer, which is the goal), and 49 μL of Tom's PCR mix.
- Run the tubes on the thermocycler at: 95deg for 3:00, then cycle through: (a) 94deg for :30 (b) 55deg for :30 (c)68deg for 2:15, then 72deg for 10:00
- Digestion - Kate [[../Notebook/2006-6-27#Restriction Digest |6-27-2006]]
- Important Notes:
- Keep all enzymes and buffers on ice for the entire process
- Want in each tube a total volume of 50 μL
- if cutting for a biobrick fabrication, use EcoR1 and Pst1 restriction enzymes (.5 -1 μL of enzyme is okay to use, just remember that too much glycerol in your digestion reaction could potentially cause unwanted star activity)
- use Dpn1 only if you are cutting a PCR product
- Digest your PCR insert and backbone separately, and also digest a control part in parallel to check that digest is working correctly
- Reaction Procedure steps: (50 μLL scale) --- in order of addition
- ?? μL Water (to make 50 μL)
- 5 μL Buffer 2 (quick vortex first)
- .5 μL BSA (quick vortex first)
- ?? μL DNA (whatever neccesary to get 800 ng -- god idea to do a fresh nanodrop of your sample and then make this calculation)
- .5 μL Pst1 and .5 μL EcoR1
- .5 μL Dpn 1 (only if digesting a PCR product)
- Post reaction steps:
- incubate at 37c for 2 hours (1 minimum and 6 maximum)
- heat shock for 20 mins at 80c to heat inactivate the enzyme, then store at 4c forever
- do a purification (elute in 30μL) just like a pcr clean up
- run a gel to check this step before moving on to ligation reaction
- Important Notes:
- Ligation - Veena [[../Notebook/2006-6-28]]
- General ligation considerations: 10μL total volume, 50ng of each DNA (but extra insert can't hurt)
- A ligation formula, to prepare each ligation reaction in a little pcr-sized tube:
- 1μL T4 DNA ligase buffer (vortex well before adding, make sure it smells strongly like wet dog!!)
- .5μL T4 DNA ligase enzyme
- 50ng (or >50 ng) of cut, cleaned-up insert...so, if we were using a cut BSMT insert that had a concentration of 25ng/μL, we'd add like 2.5μL
- 50ng of cut, cleaned-up backbone
- enough water to bring the total volume to 10μL
- The current consensus seems to be that we just leave these tubes at room temperature for about 15 minutes, and the ligation happens
- Transformation - André Transform competent cells
- Thaw ~50 μl cells (whichever you like) on ice. Do not use glass tubes, which adsorb DNA.
- Add DNA, pipette gently to mix (keep volume of DNA less than 5% of the cell volume)
- Incubate on ice for 30 minutes
- Note: If you are in a rush, you can shorten this incubation time to 5-10 min.
- Incubate cells for 50 seconds at 42°C.
- Incubate cells on ice for 2 min.
- Add 300μL of room temperature SOC (not critical)
- Incubate for 1 hour at 37°C on shaker.
- Note: Can also save some time here by reducing incubation to ~45 min.
- Note: Step can be eliminated if plating on Amp plates, but not most other antibiotics
- Spread 20 & 200 μl onto plates made with appropriate antibiotic.
- Grow overnight at 37°C.
- Sequencing - Bo
- Reconstitute primer of X nMoles by adding 10*X uL of water, making 100 uM solution (can turn into 25 uM by using 40*X)
- Centrifuge for 15 seconds.
- Add 180ng of template DNA
- Add 3.2 pMoles of primer (0.128 uL from 25 uM working solution)
- Pouring Plates - Kate
- Preparation of LB Agar
- Obtain 1.2% LB agar from the media room (500 ml per bottle).
- Be sure to sign it out
- Melt in microwave:
- loosen the cap a lot, lay down paper towels in case of spill, use 50% power (enter time, press Power, 5, Start)
- monitor as you melt, takes approx. 10 minutes per bottle (if doing 2 or 3 bottles simultaneously, add 5 minutes)
- If too hot, let the melted solution cool so that it's warm, but not hot to the touch - you should use the water bath to do this (50-60 degrees); this won't let it resolidify.
- Obtain 1.2% LB agar from the media room (500 ml per bottle).
- Once it's cool enough (50-60 degrees) - then add antibiotics: final concentration, 50ug/ml AMP; 20ug/ml KAN. (concentrations of liquid stocks: Amp; 50mg/ml. Kan, 10mg/ml.)
- For 500mL of LB Agar use:
- 500μL Amp stock
- 1mL Kan stock
- 750μL Gent stock
- 500μL Tet stock
- 500μL Chl stock
- Swirl to mix; try not to make many bubbles.
- For 500mL of LB Agar use:
- Actual Pouring
- Obtain a container of empty plates. One bottle (500ml) of LB Agar will make about one container of plates (20 plates).
- Cover the base of the plate, and then just a bit more after that.
- Recap each plate upon pouring. If there are lots of bubbles in your plates (i.e., more than one or two on the edge), you can flame the plate using the small bunsen burner to eliminate bubbles. Another way to remove the fine bubbles that may be in your flask before puring is to mist the inside of the flask with a 75% ethanol spray bottle.
- Leave plates to dry and cool for a while on the benchtop (overnight even).
- It is a good idea to label the stack of plates to indicate antibiotic.
- Store the plates in their original bags - upside down, so that the gel is hanging downwards (this keeps condensation off the gel).
- Label the bags following taping rules:
- Red tape in back (indicates LB)
- Tape in front indicates antibiotic (green=Kan, yellow=Amp, more taping/color rules are on the refrigerator in 68-564)
- Also write name of antibiotic, and concentration, on the front piece of tape.
- Store the labeled bags of plates in the fridge
- Label the bags following taping rules:
- Preparation of LB Agar
- Autoclaving Waste- Stephen
- From the waste container, remove the double-bagged waste.
- Receive one of the purple vials from Jason to place inside the waste bag. Be sure to tie it to a string such that the string can be pulled out of the bag. This will allow you to remove the vial from the bag without touching it directly.
- Place the waste into an autoclavable plastic container and take it to the kitchen's autoclave on the cart.
- On the options on the autoclave, select the liquid, default, and 1 hr settings.
- Fill out the proper information in the autoclave log binder in the far lab (closest to Endy's office) above the sink.
- Once the waste is completely autoclaved, remove the purple vial by pulling the string. Bring the purple vial to the room directly below the kitchen on the 2nd floor. On the wall (the right, perhaps?), should be a place to put the vial. Then, fill out the proper information on the notebook on the wall next to the vial plastic container.
- Tape the top of the double-bagged waste to seal it.
- Tie onto the top of the double-bagged waste a biohazard tag.
- Place the waste into the garbage barrel.
- Pouring/running gels - Veena
- First, you have to pour the gel:
- Get an empty small flask, and the 1% agarose from the 70-something degree incubator by the door (watch out...the agarose is hot. You might want to wear gloves)
- Flame the bottom of the flask to heat it up (for maybe 5-10 seconds? Not too long! the point of this is just to make sure that the gel doesn't solidify in the beaker before you pour it)
- Pour about 40mL of the 1% agarose into the flask; put the agarose back in the incubator
- put on gloves and take the flask containing the 40mL of agarose to the gel room
- carefully add 2μL of Ethidium Bromide to the agarose, making sure to keep the vial of EB in its plastic container; swirl the flask gently to mix the EB
- press a small plastic gel holder into the long plastic u-shaped thing (hard to describe...you'd probably have to see this demonstrated), and place a purple plastic gel-well-making thing in the top slot (put it in with the side with the fat purple things pointing downward)
- pour the agarose + EB from the flask carefully into the gel-holder. Try to avoid making bubbles, and make sure the agarose isn't gloppy.
- wait 20 minutes for the gel to solidify
- General note: you want to try to work quickly, since you don't want the gel to solidify before you pour it
- Next, you have to add the samples to loading dye to prepare them for loading
- First, write out the samples you'll be running on a post-it note or something, so you know exactly what order they'll be in
- Cut a piece of parafilm
- Get out the loading dye and ladder (for now, I think we'll be using the 2-log ladder) from the 4deg freezer
- Now make dots of loading dye in a row on the paraflim. Considerations: (1) you know the concentrations of the DNA you'll be running...you want to try and run at least 100ng of DNA in each well, so bands will be visible (2) if you're using 6x loading dye, you'll add 5 parts sample to 1 part loading dye, and if you're using 2x loading dye, you'll add 1 part sample to 1 part loading dye Generally, I'd use 5μL of loading dye + 5μL of sample if I'm using the 2x loading dye, and 1μL of loading dye + 5μL of sample if I'm using the 6x dye
- now you have dots of loading dye on the paraflim...so add the samples you want to run. Make sure the ladder is the first dot, and that you stick to the order you wrote on the post-it note. In order to add a sample, gently add your sample to a dot of loading dye and pipet up and down to thorough mix your sample with the loading dye (the loading dye basically lets you keep track of your gel so it doesn't run too long, and it has glycerol so your sample sinks into the well when you load it)
- Now you should have a row of blue dots on paraflim, ladder first, ready to be loaded!
- Loading the gel
- After your gel has hardened, place the gel in a gel box (like U God or inspectah deck) with the wells on the left side. If the gel isn't covered by buffer, add enough 1x TAE buffer to the box to cover the gel (but don't overfill the box)
- Load the samples carefully. Pipet up the blue dot of DNA + loading dye from the parafilm and then eject it into the well; try not to poke holes in the gel. This picture is pretty good:
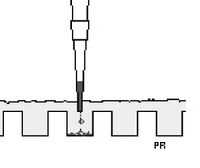
Loading a gel - Now make sure the leads are properly connected, slide the lid on top of the gel box, and run the gel with a voltage of around 85V.
- Run the gel until the blue band is about 3/4 of the way down the gel...you can take it out earlier if you're pressed for time
- Imaging the gel
- after the gel has run, switch off the machine and slide the lid off the gel box
- You should be wearing gloves anyway, but, wearing gloves, lift up the gel, drain off the excess buffer, and place the gel on a paper towel.
- Slide the lid back on the gel box and carry your paper towel + gel to the imaging computer
- change your right glove...now this is your clean hand, to operate the computer with
- using your "dirty" left hand, take the gel off the paper towel and center it in the chamber (make sure it's off (green light) before you slide open the door)
- Set the light to "upper white" and center the gel
- close the door to the chamber containing the gel and set the light to "transilluminator"; hit the green button to take an image. When an image pops up, hit the red button to capture the image and turn off the light. If it's not bright enough, you can increase the exposure time.
- Go to File->Export Image and save the image as a .jpg in the iGEM2006 folder. You should also print a copy.
- Now you have to dispose of the gel. Put the gel (in its plastic holder) back on the paper towel and take it to the gel room. slide the gel into the waste container, and rinse off the plastic gel holder.
- First, you have to pour the gel:

- Making Glycerols- Stephen
- Remove 1 mL of LB culture.
- Place it into the glycerol tubes on the far right (your right) side of the 1st shelf on the lab bench closest to Barry and Jason's space.
- Place the tubes into the -80 degree freezer (one lab over from ours around Samantha) in the iGEM box (second to the bottom shelf).
