Endy:Screening plasmid/Inverter characterization/Protocols
This is a work in progress, protocol is currently incomplete
Purpose
Characterize the in vivo transfer function of a Biobricked inverter quickly and reliably.
Background
The inverters should be present in one of the PoPS device screening plasmids, this protocol refers to characterization of inverters in Screening Plasmid 1.0 in CW2553+pJat8 [1, 2]. pJat8 provides Gen resistance and the screening plasmid has Amp resistance. SP1.0 uses an arabinose induction system, so cells shouldn't be grown with glucose because of the intereference of catabolite repression.
The current characterization protocol follows the following steps:
Day 1:
- Grow the cells overnight in M9/gly to achieve density.
Day 2:
- Dilute back to return the cells to mid-log
- Innoculate experimental cultures of M9/gly/arabinose
Day 3:
- Harvest cells for FACS
- Run FACS
Procedure
Each culture has a different growth rate, so ensuring that the intermediate dilution and experimental culture remain in log phase can be difficult. Additionally, the final culture must have sufficient induction time (so that the fluorophore concentration reaches steady state) and growth time (so that the culture is dense enough to measure), while still keeping cells in mid-log. From general experience, cells with an inverter device + SP1.0 usually grow very slowly. Rough guidelines for timing are included below.
Day 1
Set up Overnights
Set up one 5ml overnight culture per experimental device or control early in the afternoon in M9/glycerol and give them plenty of time to grow up.
- Controls
- Negative Control - CW2553/pJat8
- Empty Plasmid - This provides a baseline for calibrating GFP and RFP
- GFP only (see controls) - This enables quantification of bleed from GFP in RFP filter
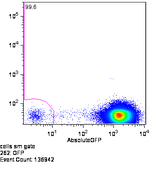
GFP under constitutive expression off of Ptet, measured on the MoFlo. Not much bleed into the RFP channel on this machine.


Day 2
Dilute back in AM
For high copy plasmids, rough doubling time is 1.5 to 2 hours. Depending on how much the cultures have grown up overnight, dilute back an appropriate amount into 5ml of M9/glycerol so that the cells will be dense enough to innoculate the experimental cultures but still in mid-log (about 100x usually). If the cultures have not reached stationary after being grown overnight, experimental cultures could possibly be innoculated from the overnight culture. However, one would ideally like to keep a consistent protocol between different constructs.
Set up overnight experimental cultures
- At least a 12-hour induction time is needed to allow the fluorophores to reach steady state (previous results with an empty screening plasmid have shown unchanged levels between 12 and 14 hours). Time to reach steady-state varies between devices, depending on the growth rate. We would like a rough minimum of 9 doublings before measurement, though this number might be greater for higher induction levels.
- It's usually safer to innoculate with a small number of cells and give them plenty of time to grow and respond to the arabinose without reaching stationary.
- The induction limits of SP1.0 range from 0% to about 0.003% (what is upper limit exactly?) arabinose. At higher concentrations the induction begins to drop off.
- Using six different induction levels (0%, 10-6%, 3x10-6%, 10-5%, 3x10-5%, 10-4%) provides a good transfer function for Q04400.007.
- Each experimental culture, as well as the empty screening plasmid control, should be tested at all arabinose concentrations.
- The negative control can be grown with no arabinose.
- The GFP only control can be grown with no arabinose if using Ptet. If Pbad is being used, grow with high arabinose.
- The total number of samples will be ((EXP+1)*#arab concentrations + 2) * #replicates
Note: It is usually a good idea to make up large batches of media + arabinose and then make 5ml aliquots of this for each experimental device -- this ensures they all see the same levels of arabinose.
Day 3
Prepare FACS Samples
Take 1 ml aliquots and place on ice.
Run samples on FACS
- Print this sheet to take along to the FACS facility.
- Remember to set the PMT voltages, SSC threshold, and SSC voltage. Flow Cytometry Center staff can also do something to calibrate the fluoresence readings based on the beads.
- The droplet maker can be turned off to reduce noise in the signal if you are not sorting the cells.
- Run beads (~6 drops in 500ul) - this is used to verify that the lasers are aligned as well as to serve as a calibration between runs, you can read more about this in the analysis techniques. Be sure to save the bead calibration data.
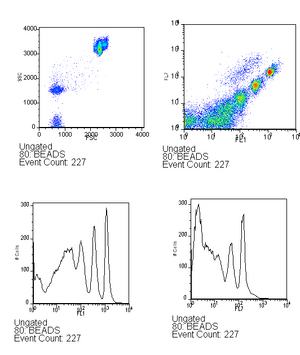
Beads should look approximately like this with good alignment. - Run the GFP control, followed by the negative control.
- This allows you to determine the "noise region", since you shouldn't trust readings that are within the range of autoflourescence of your cells. Also, it makes sure that the method being used to clear the tubing between samples is working well. If you still see many positive cells in the negative control then you can try running bleach between samples. (This is maybe more important between different devices than between different arabinose concentrations).
- Run the experimental samples. (have been collecting ~100K cells / sample)

References
- Khlebnikov A, Datsenko KA, Skaug T, Wanner BL, and Keasling JD. Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiology (Reading). 2001 Dec;147(Pt 12):3241-7. DOI:10.1099/00221287-147-12-3241 |
- Horazdovsky BF and Hogg RW. Genetic reconstitution of the high-affinity L-arabinose transport system. J Bacteriol. 1989 Jun;171(6):3053-9. DOI:10.1128/jb.171.6.3053-3059.1989 |
Contact
- protocol based on screening plasmid protocols developed by Josh Michener

{kind=link}