Calcium is the most abundant mineral in our body yet the concentration of Ca2+ within our cells is extremely low (~10-7M). Calcium pumps and gated channels work hard to establish and keep this steep gradient. Inside the cell, calcium is spatially restricted, stored in some but not all organelles, poised for release when appropriate. Calcium-handling proteins are a large and widely distributed family of proteins. Many share a characteristic dumbbell shape, with two globular domains connected by a flexible linker region. You'll remember calmodulin, the calcium sensing protein whose structure you examined last time, that binds four Ca2+ ions, two with high affinity and two with low affinity. The high affinity sites can be filled at low calcium concentrations, but when calcium gets released from intracellular stores, the low affinity sites are also filled, inducing a large conformational change in the protein. The change exposes non-polar regions of calmodulin that can bind to non-polar regions of a target molecule. Notice how the backbone of calmodulin extends in the presence of calcium (figure from the review by Vetter and LeClerc) in FEBS 2003.
calmodulin's shape with (right) and without (left) Ca2+, from FEBS 2003
An interesting "just so" story related to calmodulin is the mode of action for anthrax toxin, which exploits both the cellular distribution of calmodulin as well as its mechanism for calcium sensing. One component of the anthrax toxin, called EF, can bind to calmodulin in the “low calcium” conformation, twisting the linker so it cannot respond to high calcium concentrations, effectively blocking its natural signaling functions. With calmodulin out of the picture, the cells have no way to convert the cAMP formed by EF back into ATP, leading to depletion of the cell’s energy stores. This mechanism seems particularly clever when you remember that calcium-sensing motifs are widespread and abundant but only in animal cells. Thus the Bacillus anthracis bacteria don’t deplete their own energy stores when anthrax toxin is expressed.
zircon structure
Proteins are not the only means of "handling" calcium. Dyes that change their spectral properties are also available and find particular usefulness as a direct read-out for free Ca2+ in solutions or inside cells. Water hardness tests are one example where these dyes are used, relying on a dye called "Zircon" (structure shown) which changes its color when it reacts with minerals. Calcium and magnesium are the principle minerals that make water "hard," though water can have other minerals like strontium and barium that will also react with Zircon. It turns out that Zircon is also one of the active ingredients in dandruff shampoo, though its spectral properties probably aren't so relevant in that application since most people close their eyes when they shampoo!
Structure of Fluo-3, pentammonium salt form
Since the concentration of free Ca2+ is so very low inside cells, other dyes have been developed that can measure in the submicromolar range. For the most part, these dyes increase their fluorescence in the presence of calcium. The dye we will use, Fluo-3 (structure shown), can be excited by light at 506 nm which is in the visible range. The dye binds Ca2+ with a Kd around 800 nM (this measurement naturally depends on temperature, pH, ionic strength etc), increasing the dye's emission at 526 nm by more than 100-fold, as described. The sensitivity of Fluo-3 and other dyes like it (Fura-2, Indo-1...) make them useful for measuring free Ca2+ inside cells. In vitro, however, it's tricky to make solutions with such low concentrations of free Ca2+. The most reliable method is to nearly balance the solution's concentration of calcuim with a chelator, namely EGTA. Under these buffered conditions, the free Ca2+ in solution depends on the dissociation constant of Ca2+ from EGTA and can be calculated as:
[Ca2+]free = Kd for EGTA x {[CaEGTA]/[K2EGTA]}
Today we will measure calcium concentrations of several solutions. Beyond emphasizing some important laboratory techniques (like standard curves and spectrophotometric analysis of chromogenic reactions), today’s work should underscore the limited sensitivity and range of such techniques. If you remember only one detail from today's work, let it be the importance of choosing an appropriate tool for the measurement you want to make.
Luckily cells are better at sensing calcium that we are in the lab, as we’ll see when we begin measuring Ca2+in vivo using a genetically encoded calcium sensor. Today you will prepare the MES cells you'll need for that experiment next week.
Protocols
Part 1: In vitro Ca2+ measurements
We will compare two methods for in vitro Ca2+ measurements, one that relies on changes in a chromogenic compound that can be measured spectrophotometrically and another that relies on fluorescent changes of a sensitive indicator dye.
Assay 1: chromogenic
We will be using a calcium assay kit sold by "BioAssay Systems" (catalog #DICA-500) that's more commonly applied to diagnose kidney, metabolic and blood disorders. Samples (urine, saliva, serum or in our case CaCl2 solutions) are mixed with a dye that forms a blue color in the presence of free Ca2+. The intensity of the blue color directly reflects the concentration of Ca2+, within the linear detection range of the kit. This range is reported to be 20 uM to 5 mM. You will directly measure the linearity of the assay within this range and beyond. Then you will determine the concentration of free Ca2+ in solutions that are relevant to your tissue culture experiment.
Make 50 ml of a CaCl2 stock solution (Fw 110.99) that is 4X more concentrated than the highest value stated as linear in the BioAssay Systems kit. Use only the good water from the back room of the lab since this will not have free calcium contaminating it. Alternatively, you can try to use the tap water as a point of comparison to others in the lab. It might be interesting to know just how much Ca2+ you can measure this way.
Dilute this stock in 2X steps, using a 96 well dish or in eppendorf tubes. You should make 100 ul of each dilution. Stop when you are 4X lower than the lowest concentration stated as linear in the BioAssay kit. Be sure to mix each dilution before making the next in the series, and change tips between dilutions.
Move 5 ul of each dilution to a cuvette, noting which cuvette has which dilution.
You should also set up three cuvettes, each with 5 ul of a solution relevant to the mouse embryonic stem cells. This could be Growth Media, or Trypsin solution, or OptiMeM or whatever. Alternatively, request an unknown from one of the teaching faculty.
Set up a table in your notebook to record the spectrophotometric data you will collect. The table should include a row for sample descriptions and another for the absorbance values you'll measure at 612nm. You could also include a column to record any observations you have of any sample (color, clarity etc).
In a 15 ml conical tube, mix 5 ml of BioAssay Reagent A with 5 ml of BioAssay Reagent B.
Add 500 ul of the assay mixture to each cuvette and mix by inverting several times using a strip of parafilm or a gloved finger to cover the top of the cuvette.
Incubate at room temperature for at least 3 minutes. Samples are stable for more than an hour.
Use 500 ul of assay mixture to blank the spectrophotometer (612 nm) and than record each sample you have prepared.
Enter the data you've collected into an Excel spreadsheet. Then:
prepare a graph with all the standards you measured. What is the equation for the best fit line? What is the R-squared (a measure of linearity). Print out a graph for you lab notebook.
prepare a graph with only the concentrations of CaCl2 that fall within the stated linear range of the BioAssay Kit. What is the equation for the best fit line? What is the R-squared value? Print out a graph for you lab notebook.
Using the best fit line you most trust (state which and why you chose that one), determine the concentration of calcium in the cell culture solution you chose. How well do the triplicate measurements agree with one another?
Enter the requested information onto the discussion page associated with today's lab.
Assay 2: Fluo-3, salt
Nanodrop Fluorimeter
We will titrate the concentration of free Ca2+ in solution by mixing different amounts of K2EGTA and CaEGTA. The reactions of these solutions with Fluo-3 dye will be at room temperature, pH 7.2 and 100 mM KCl. Under these conditions, the Kd for EGTA is 150.5 x 10-9 M. Measurements of Fluo-3 fluoresence will be made on a Nanodrop Fluorimeter. Please be extremely careful with this instrument. The lab has only one and, though it looks like a toaster, it costs approximately $10,000.
1. Begin by mixing the relative volumes of K2EGTA and CaEGTA according to the following table.
Tube number
Volume K2EGTA, ul
Volume CaEGTA, ul
Calculated free Ca2+, uM
RFU
zero (blank)
1000
0
0
0
1
900
100
0.0167
2
800
200
3
700
300
4
600
400
5
500
500
6
400
600
7
300
700
8
200
800
9
100
900
2. Calculate the concentration of free Ca2+ in each solution using the formula
[Ca2+]free = Kd for EGTA x {[CaEGTA]/[K2EGTA]}
and a Kd of 150.5 x 10-9M. To help you get started, the first calculation has been done.
3. Add one ul of 1 mM Fluo-3 to each solution including the blank and flick to mix.
4. Read the fluorescence of each solution on the Nanodrop by
attaching the nanodrop through the USB port of your laptop
opening the ND-3300 software
selecting "Fluorescence Profiler" from the menu that appears
reading and recording the "Peak RFU" value for the blue source.
5. Blank the nanodrop by placing 1 ul of the K2EGTA blank on the lower sensor and gently lowering the arm to meet it. You do not need to push the arm down for the sensors to meet. The nanodrop will do this automatically when it makes its reading.
6. Select "blank" to zero the machine using this solution.
7. Wipe the top and bottom sensor with a water soaked Chemwipe.
8. Dry the top and bottom sensor with a dry Chemwipe.
9. Measure the relative fluorescence units (RFUs) associated with the 0.0167 uM solution by placing 1 ul of this solution on the bottom sensor and gently lowering the arm.
10. Select "Measure" from the upper left corner of the program screen. The measurement will appear in the lower right corner. You should record the Peak RFU value for the blue light source in a table in your lab notebook and/or on the wiki.
11. Repeat the two Chemwipe cleaning of the sensor and measure the remaining samples, using the "back" button to prepare the instrument.
12. If you have time, plot the RFU vs [Ca2+]free using Excel. You will be asked to post your plot as part of your "for next time" assignment.
Part 2: Cell culture
We will spend another part of lab today in the cell culture facility, moving MES cells to slides. This protocol is quite similar to the one from last time (so if you need reminding, refer back to the directions from last time). There are two major differences in today's protocol. One is that you will resuspend the cells in media that lacks antibiotics. This is essential to prepare the cells for transfection, but it puts your cells at additional risk for contamination. You'll have to use your very best asceptic technique. Another important difference today is that you will be growing the cells on slides rather than in flasks or dishes. This will allow you to better observe the cells later in this experimental module with the fluorescent microscope.
4-well slide
You and your partner should each prepare a slide, though you can share the cells from one flask.
Begin by preparing the cell culture hood as you did last time.
Pregelatinize each chamber of a 4-well slide, using 0.5 ml of gelatin dispensed from a 2 ml pipet.
Retrieve one flask of cells from the incubator.
Aspirate the media.
Wash the cells with 5 ml PBS.
Add 2 ml of trypsin for one minute precisely, aspirate the trypsin, then incubate the "dry" flask in the 37°C incubator for ten minutes precisely.
Triterate the cells with 5 ml "pre-transformation media."
Dilute the cells 1:20 in pre-transformation media. You will need only 2 ml of dilution per slide. You and your partner can share one dilution mix.
Aspirate the gelatin from each well of the slides you've prepared.
Aliquot 0.5 ml per well.
Place your slide in a petri-dish that can be returned to the incubator until next time. One of the teaching faculty will feed your cells the day before you return to lab.
DONE!
For next time
If you didn't have time to finish in lab, add your in vitro Ca2+ measurement data to the table that is on the "discussion" page associated with today's lab.
If you didn't have time to finish in lab, post a thumbnail graph of the Fluo-3 RFU vs [Ca2+]free data you collected on to the "discussion" page associated with today's lab. Be sure to list your team.
Next time you will transfect the cells you plated today. Timing and concentrations are very important for this part of the experiment. Consequently, you should read ahead to the protocol of the next lab. Specifically you should know
how much Lipofectamine you will add to how much OptiMEM for each transfection and for the transfection cocktail
how many of the slide's chambers you will "mock" transfect, and how many will be transfected with the "inverse-pericam" plasmid
how much DNA you will add to how much OptiMEM for each transfection and for the transfection cocktail
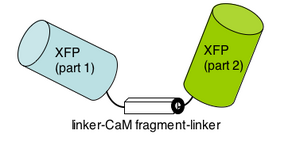
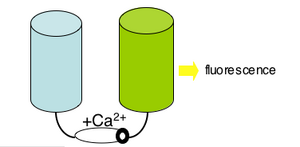
Please read the protein design work described in the 2004 PNAS article by Nagai et al. The "cameleon" protein they describe is very much like the genetically-encoded calcium sensor you will use next time. apo cameleon These proteins use fluorescence as a readout for calcium ions. The cameleon is a clever chimeric fusion protein that links the calcium responsive portion of calmodulin (as well as a portion of myosin light chain, confusingly named M13 of all things!) to a fluorescent pair. Ca2+ cameleon The cameleon's "apo" form (no Ca2+) holds the fluorescent molecules in a conformation that generates no fluorescence. When the cameleon binds Ca2+, the calmodulin domain "scrunches" and brings the two parts of the fluorescent molecule together.
Different variations of this intelligently designed sensor exist, including some that are "FRET" based (using two full fluorescent proteins and relying on a transfer of energy from one to the other), and some that "divide" a single fluorescent molecule, requiring the calmodulin "scrunch" to bring the halves together. The latter is what we'll be using in lab next time. If you'd like to see these sensors in action check out the quicktime movies that are associated with the 2004 publication we will discuss next time.
Here are some questions to focus our discussion of the Nagai paper. Please choose one question that you will be the "point person" for and put your (initials/lab section/team color) next to that question so no one else claims it.
What is “Venus,” and what does it mean for this protein to “mature”?
Why is circular permutation of GFPs part of a strategy for improving the cameleons?
What determined where new N- and C-termini were created for the circularly permuted Venus variants?
Why are the fluorescence spectra of the calcium free YCs so similar to one another (dotted lines in Fig. 1C)?
What are fluorescence anisotropy and steady state polarization?
What factor(s) may cause differences in the calcium titration curves for YC2.60, YC3.60, and YC4.60 (Fig. 2B)?
Why is the pH dependence of the fluorescence changes important?
Why is fluorescence generally shown as a ratio between values for two wavelengths?
In Fig. 3, why does the fluorescence emission of ECFP seem to vary inversely with the emission of cp173Venus?
What are [Ca2+]c and [Ca2+]pm, and why might [Ca2+]pm be bigger?
By what methods were changes in intracellular calcium concentration induced?
Why is having a larger dynamic range an important advantage for the new YCs?
In addition, please check out the following to help prepare for the discussion:
Some acronyms to know: GFP, YFP, CFP, CaM, YC, FRET, cpGFP

-S07_class_specific_info-wiki_art-apo_vs_Ca_bound_from_FEBS.png)