BISC314:Full Protocol

Laboratory Protocols
LAB #1: Learning Sterile Technique and Field Trip to the Cheese Shop
Today we will be taking a trip in to Cambridge to visit Formaggio Kitchen, a famous cheese shop in the area - they even have a cheese cave! Each of you will pick a cheese to be your microbial habitat for the next few weeks. We will sample a large variety and learn about where these cheeses came from. Let's be sure we have representative cheeses from these four main classes:
1. A blue cheese
2. A fresh cheese
3. A washed rind cheese
4. A soft, brie-like cheese
Back in the lab, we'll review sterile technique and inoculate media of various kinds from our cheese rinds. We will isolate a beautiful array of different microbes (both eukaryotic and bacterial) from these cheeses and in the next few weeks you'll be using them to investigate two major microbial community functions: growth interactions and chemical signaling. If you haven't taken BISC209, or if you feel rusty on your microbiology, you might want to peruse the following link: BISC209:Protocols
Streaking out our cheese isolates
1. Make sure to wear gloves and use proper sterile technique throughout
2. Get 4 NB agar plates and label them with your name, the date, your cheese, and what part of the cheese you are inoculating from
3. Flame your loop to sterilize it
4. Pass your flamed loop over the surface of the cheese - you don't need a chunk! Microbes are invisible - don't get a big scoop of the cheese on your plate as this will lead to over-growth
5. Use your loop to smear out this inoculum over about 1/4 of the plate. Use the image below to help guide you.
6. Reflame the loop and use it to dilute this smear further. Pass the loop through the initial, most dense streak once. Then make a second smear on the plate using the loop.
7. Continue till you've completed the pattern below on all 4 plates
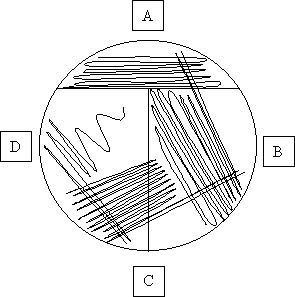
8. Place your NB plates agar side up on your bench. We will come back Friday to check on them
LAB #2: Macroscopic and Microscopic observation of Isolates
You will have many different colonies growing up on your plates from last week. In your lab notebook, take some time to look at your colonies and describe their morphology, color, and smells! Does your Camembert inoculum smell like Camembert? You may find the following link useful for colony morphology descriptions: ASM MicrobeLibrary
Let's also look at our isolates under the microscope. We will make smears of our organisms next. Before we get to that point, however, it's worth discussing cellular morphology a bit. For the most part, bacteria are much smaller (0.2 to 4 µm) than eukaryotes (~100 µm). We will be using the 100x objectives to see bacterial morphology under the scope. You may be able to see the shapes of many eukaryotes you've isolated under 40x magnification.
Background
Bacteria come in many different shapes (see this Wikipedia article for a nice figure depicting different shapes). These shapes are not, however, good indicators of relatedness or even species type. Many bacteria can be multiple shapes (termed pleomorphism) depending on how you grow them or from what conditions they are isolated. However, traditional names for bacteria often elude to their shape: for example, Vibrio fisheriis a curved (vibrio) bacterial organism.
You will make smears of all of your isolates.
Making smears of your isolates
1. Label a clean, glass slide with a graphite pencil on the far left of the slide with the code name of the isolate. For example, my 2nd isolate will be my initials followed by the number 2 (IN-2)
2. Place a small loopful of deionized water on the slide as far from each other as possible.
3. Flame the loop, allow it to cool for a few seconds and touch the cooled loop to a colony of your isolate, picking up a TINY bit of white growth from the bacterial colony. An invisible amount of growth obtained from just touching the cooled loop to the colony is fine.
4. Place the loop with the bacterial growth into the drop of water on the slide. Use a circular motion to make a smooth suspension of the bacteria in the water. Stop when there is a circle of emulsified bacteria about the size of a nickle on the slide.
5. Reflame the loop.
6. Add a coverslip to your slide and take it over to the microscope -it is ready for viewing!
Lab Questions:
1. First, look at all of your isolates and determine if they are bacterial or eukaryotic. Can you think of a way to test if you are correct?
2. In your lab notebook, note the shape and size of each isolate.
3. Is your organism swimming around or are you observing brownian motion? How can you tell? Could you think of a way to test for motility?
The Gram Stain
Bacteria have been classically divided into two main groups based on a simple staining protocol: the Gram stain. It is named after its inventor (hence the capitalization) and is used primarily in clinical microbiology. The importance of the Gram stain is that it can distinguish many bacteria based on the architecture of their cell walls. However, not all bacteria can be classified by the Gram stain and many are what is known as "Gram variable" depending on what stage of growth they are in. Gram-positive and Gram-negative bacteria are both stained purple by the crystal violet (primary) stain. Addition of the iodine leads to the formation of a crystal violet-iodine complex within the cell wall. The decolorizer extracts lipid from the cell wall of Gram-negative bacteria, so the crystal violet-iodine complex diffuses from these cells and loses color. The crystal violet-iodine complex remains within the Gram-positive bacteria because their cell walls are think and lack the lipid-rich outer membrane of Gram-negative bacteria. Due to the increase in porosity of the Gram negative cells after lipid loss from decolorization, safranin (counterstain) is able to permeate the cell wall of Gram-negative bacteria. Purple (primary stain retaining) indicates Gram positive and red (counter stain uptake) indicates Gram negative. Some organisms and dead or dying cells do not take up or lose the stain appropriately and can not be classified as either Gram positive or Gram negative.
1. Label a clean, glass slide with a graphite pencil on the far left of the slide with the code name of the isolate. For example, my 2nd isolate will be my initials followed by the number 2 (IN-2)
2. Place a small loopful of deionized water on the slide as far from each other as possible.
3. Flame the loop, allow it to cool for a few seconds and touch the cooled loop to a colony of your isolate, picking up a TINY bit of white growth from the bacterial colony. An invisible amount of growth obtained from just touching the cooled loop to the colony is fine.
4. Place the loop with the bacterial growth into the drop of water on the slide. Use a circular motion to make a smooth suspension of the bacteria in the water. Stop when there is a circle of emulsified bacteria about the size of a nickle on the slide.
5. Reflame the loop.
6. Allow the liquid on the slide to dry.
4. Be sure all the liquid on the slide has evaporated before proceeding to heat fixation (or you will explode the cells in the next step).
5. Heat fix (to kill and attach organisms to slide) by rapidly passing the slide (smear side up) through a flame 3 times. Use a slide holder and avoid contact with hot glass.
To Gram stain the desired organism(s):
Use the staining trays and sink area. Be sure you evenly cover all the smears on the slide throughout this procedure.
1. Place your smear on the staining tray. It is important that the slide be level during staining so use paper towels under the tray to get it leveled. If you do, it is much easier to prevent dye from running off the slide during the staining process and to be sure that your smears are evenly covered with each reagent.
2. Dispense just enough Crystal Violet solution (0.5% crystal violet, 12% ethanol, 0.1% phenol) to completely cover each smear and stain for 1 minute. (Crystal violet is the primary stain.)
3. Rinse the slide by lifting it at a 45 degree angle (using gloves or a clothes pin or slide holder) in a very gentle stream of water that is directed above the top smear until the waste water coming off the bottom is relatively clear; drain off excess water by touching the edge of the slide to a paper towel.
4. Dispense just enough Gram's Iodine (mordant)to completely cover each smear. Let stand for 1 minute, and rinse thoroughly with a gentle stream of water as in Step 1.
5. Lift the slide at a 45 degree angle and drip Decolorizing Reagent (80% isopropyl alcohol, 20% acetone) down the length of the slide making sure it comes in contact with all three smears. This step is tricky as it is easy to over- or under-decolorize. Do this for 10 seconds and IMMEDIATELY rinse, as in step 3, with a gentle stream of water.
6. Place the slide flat on the staining tray and dispense just enough Counterstain Solution (0.6% safranin in 20% ethanol) to cover each smear. Let stand for 2 minutes; rinse with water as in step 3.
7. Blot dry using the bibulous paper package found in your orange drawer. Do not tear out the pages, just insert your slide and pat it dry.
8. Clean up your area; rise your staining tray. Leave it upside down by the sink on paper towels
9. Observe your stained microbes microscopically following carefully the procedure for using the the oil immersion objective on your compound brightfield microscope BISC209: Microscopy described in the protocol pages of BISC209.
Preserving DNA from your isolates
For each of your isolates you should save a small amount of DNA for later identification. The protocol is below:
1. Flame the loop, allow it to cool for a few seconds and touch the cooled loop to a colony of your isolate, picking up a TINY bit of white growth from the bacterial colony. An invisible amount of growth obtained from just touching the cooled loop to the colony is fine.
2. Place the loop within a 1.5 ml eppendorf tube containing 200 µl of water.
3. Vortex the tube to fully resuspend the isolate.
4. Freeze the tube at -20C for later. We will be identifying the most interesting isolates using molecular methods after our lab assays
Special Field Trip: Woods Hole
On Sunday, September 26th, we will be traveling to Woods Hole to meet the Marine Biology lab and see sulfur metabolism and Cyanobacteria in their natural environment. We will be meeting in Gray parking lot at 10AM - bagel breakfast + coffee provided. It will take us 1.5 hrs to get to Woods Hole and meet the Marine Biology class. We will have lunch there (also provided). While at WH, we will explore tide pools and learn about microbial mats and how animals survive in sulfur-rich habitats.
LAB #3: Antagonistic and Mutualistic Interactions
- NOTE: You must remember to set up overnight cultures for 8 of your isolates the prior Friday!
The microbial community living within the cheese is a complex one with many different microorganisms. As is true of any environment, these microbes interact with each other - both metabolically and chemically. Today, you will be taking your isolates and testing them against each other for growth inhibition or growth benefit. Some of these organisms may prevent the growth of others through the production of chemicals we'll discuss in lecture; others might promote the growth of their neighbors by secreting metabolites that are needed.
Interaction Assay
1. Create a 96-well template for your isolates. Using the excel template found here Media:template.xls make up the map for this 96-well plate - you should know where each of your 8 chosen isolates will be.
2. Using sterile technique, take 50 µl of your isolate from the overnight culture and place it into the appropriate well. Add another 100 µl of sterile media to the well to dilute the culture
3. When you are finished loading the top and side wells, use the p 20 micropipette to move 10 µl from wells A2 - A8 into wells B2 - B8. Repeat this until H2 - H8 has 10 µl in it also.
4. Now we will add the organisms from wells B1 - H1 to the rest of the plate. Again, using the p 20, take 10 µl from wells B1 - H1 and mix into the wells B2 - H2. Repeat this process until you have mixed all of the possible combinations of your isolates on this plate.
5. Each of your wells now has isolates growing by themselves and isolates mixed together. We will inoculate from this plate onto a square agar tray in order to observe what these organisms look like when grown together. For this we will use the multichannel micropipette. Using the multichannel, take 5 µl of your inocula and deposit a drop on top of the square agar tray. Repeat this until you have completed the full array.
6. Wait for your spots to dry before placing your trays upside down.
You should come in to the lab a few times this week to check on your assay and note any interesting results
LAB #4: Quorum sensing - chemical signaling within our community
Many bacteria are able to secrete signals into their environment that they can then utilize to determine their own density. Since bacteria are single-celled organisms, why do you think they would they be interested in knowing how many of them are near? A very well studied example of the quorum sensing system comes from Vibrio fisheri, a bacterium that produces light only at high densities. If you think about it, the light produced by a single bacterium is unlikely to be seen by anyone so Vibrio wait until they have reached a "quorum" before turning on the metabolic pathway that creates light. In this way, a gene regulatory network is actually controlled by cell density. To hear more about it from another source, see this YouTube video of Bonnie Bassler.
Today we will be testing if any of your isolates are secreting Auto-Inducer (AI) into the surrounding media. Specifically, we will be using a strain of bacterium called Chromobacterium violaceum. This organism is gram-negative and normally found in the soil. It produces a very strong purple pigment (hence the name) in response to levels of AI. We will also use a violacein-negative, mini-Tn5 mutant of C. violaceum (CV026) which can produce pigment in response to the AI from other bacteria but can no longer secrete its own AI - this will be our biosensor.
1. Label your agar plates. You will draw a line down the middle of the plate and write CV026 on one side and your isolate code on the other side. Also label a plate for our positive control, the parent strain to CV026: Chromobacterium violaceum ATCC 12472.
2. Flame your loop. Use your loops to dip into an overnight culture of CV026. Streak 1/2 of an agar plate with this organism (the side with the CV026 label).
3. Flame your loop. Grab a relatively large amount of your isolates and streak them near to the CV026 streaks. Re-flame your loop.
4. Dip the loop into an overnight culture of ATCC 12472 and streak it near to the CV026 streak.
5. Place your plates upside down till next week. If your isolates are producing AI sensed by CV026, you will see a purple pigment.
Recreating your cheese rind -- isolates to community
Today we will also be attempting to recreate the rinds found on your cheese by inoculating a fresh cheese (chevre) with your isolates in combination. You can set up three different experiments and observe what the rinds look like over the next few weeks. Choose three different combinations of isolates or those from different parts of your cheese.
1. Label an empty petri dish with the experimental condition.
2. Label a 1.5 ml microcentrifuge tube your isolate designation and add 200 ul of water to the tube.
3. Flame your loop. Use the loop to take half of an isolate patch and resuspend it in the microcentrifuge tube.
4. Repeat steps 2 and 3 for each of your isolates.
5. Label a 1.5 ml microcentrifuge tube for each experimental condition.
6. Using a micropipette, take 50 ul from each isolate to mix into the experimental condition. For example, if my experimental condition #1 was isolates 4-6, I'd take 50 ul of isolate 4, 50 ul of isolate 5 and 50 ul of isolate 6 in one tube labeled condition #1
.
7. Make sure your isolates are well mixed in the tube.
8. Take a slice of the chevre and place it into the petri dish.
9. Using a micropipette, take 200 ul of your experimental mix and spot it onto the top of the chevre.
10. Close the lid on the petri dish and let the cheese age. We'll keep an eye on them week by week.
LAB #5 and field trip #2: the dairy
Today we will travel on our second field trip - to the Oak Knoll Dairy. There we will hear about how they raise their cows and how they process their milk for cheese making. When we return, we'll use the LAB media to select for Lactic Acid Bacteria in the milk and start our yogurt. LAB's will be cultured at 37 C on MRS media
Yogurt-making
- Note, you must come in the day after to place your yogurt in the 4C room!
Yogurt is produced by the metabolic activity of bacteria. The consistency of our yogurt will be determined by the addition of powdered milk (which thickens the yogurt) and the acidification of the milk by the lactic acid bacteria naturally found in the milk.
1. Place 250 ml of milk from the dairy into a 500 ml flask.
2. Add 4% (weight/volume) of powdered milk (10 grams in 250 ml) and blend it into the milk by swirling.
3. Heat the mixture in a water bath to 85C for 20 minutes
4. Cool the milk to 40C in an ice bath. You can tell it's cooled enough because it will feel warm to the touch, but will not be unpleasant to touch
5. Add a hefty spoonful of our yogurt starter culture aseptically
6. Incubate at 40C overnight.
7. In the morning, take your 250 ml flask and place it in the refrigerator (4C)
LAB #6: Phage and LAB's
- Note: Come in the day prior to inoculate overnight cultures of your LAB's!
What are bacteriophage?
Bacteria, just like our cells, can be infected by viruses known as bacteriophage (or phage for short). Phage are mobile genetic elements that use the infected cell's machinery for protein and nucleic acid synthesis. Without a bacterium, a phage cannot replicate. For this reason, and like viruses, phage are not considered to be living things. You can read more about phage here. Phage are important players in mobilizing all sorts of genes between cells. For an interesting read on this check out this interesting paper.
Importantly, phage have two parts to their life cycle: they can exist as either lytic (where they are actively lysing their host cells) or lysogenic (where they are integrated into the genome and being passed on generation to generation). Phage that are lysogenic can be induced to become lytic under certain conditions. Could you think about what those conditions could be? What would make you want to abandon your host?
Today we will be using a drug, mitomycin C, to induce production of active phage particles from lysogenized phage in our LAB's. Your overnight cultures will serve as inocula for a larger culture of your LAB isolates. We will then add mitomycin C and measure the OD600 at 20 minute intervals. How do you think the OD600 will change if a lysogenized phage becomes lytic?
- Measure the OD600 of your overnight culture of LAB. You will do this two ways: either using the side-arm spec or by using a cuvette in the regular spec. You will use the cuvette method first, as it is more accurate for higher density cultures (like your overnight culture) but throughout your time course, you will use the side arm spec method.
regular spec use
1. Aseptically transfer 100 ul of your culture to a 1.5 ml microcentrifuge tube. Cap the tube and spin it at max speed for 1 minute.
2. Overturn the tube onto a paper towel to get rid of the media. The cells should stay as a pellet on the bottom of the tube.
3. Resuspend the sample in 1 mL of PBS by pipetting up and down and then pipette the sample into a labelled cuvette. Make up a blank cuvette as well with PBS alone.
4. Take the sample over to the spectrophotometer and measure the OD at 600 nm.
5. Remember - your sample is a 1:10 dilution of your original culture! Multiply the number you get from the spec by 10 to get the true OD600 / ml.
side-arm spec use
1. The side arm spectrophotometer takes a measurement of OD600 through the side arm of the flask. This makes it easy to measure the OD600 without contaminating your culture.
2. Insert the culture tube in the spectrophotometer. Take note of the measurement as it appears on the instrument.
Beginning the experiment: Mitomicin C treatment
Calculate how much of your overnight culture you will need to create an OD = 0.2 culture of your LAB's in 10 mL. You can use the calculation below:
10 mL x 0.2 OD = X mL x ## OD600 O/N culture
1. Aseptically add the volume necessary of your O/N culture to two 10 mL culture tubes containing fresh LAB liquid media. One of these you will label as your experimental tube and the other will be your control.
2. Aseptically add mitomycin C to your LAB experimental culture. Note the time in your lab notebook and set a timer for 20 minutes.
3. Measure the OD600 of your 10 mL cultures every 20 minutes for 2 hours. You'll fill in the table below:
| Isolate Code Name | Time | OD600 |
Plot the OD600 of both of your cultures (your experimental and your control) using excel. Did the mitomycin treatment result in a different rate of growth for your cultures? A drastic reduction in OD600 means that you successfully created phage particles from a lysogenized phage in your LAB genome. Now the question is: is your phage relevant to its environment? What is its host range? In the next lab, we will determine the specificity of your phage by testing it against important bacteria used in yogurt (Lactobacillus species).
if you did get phage particles, let's save them by placing your cultures in the fridge (4 C) until next time.
LAB #7: Phage specificity
Today we'll be testing the specificity of the phage you isolated last time we met. Sherly has grown up overnight cultures of important organisms used in yogurt making. The question is, are your phage able to infect these alternative hosts? We will test this by using the double agar method. You will first filter out your host from last week to isolate the phage. These phage will be mixed with a small amount of the cultures provide by sherly, plus calcium chloride and molten agar. They will be poured over a hardened layer of agar. The bacteria grow within the agar and the phage, if able to lyse the host, produce circular plaques in the agar - basically wherever bacteria are missing. For cool pictures of bacterial plaques look here.
1. Filter your 10 mL culture presumed to include bacteriophage using a syringe and a syringe filter (0.2 um). You can do this by first suctioning up 5 mL into the syringe aseptically. With the plunger in the syringe, screw the 0.2 um filter into the end of the syringe. Put the tip of the syringe/filter into a 15 mL falcon and depress the plunger slowly. The drops that make it through the filter should be phage particles.
2. For each important host to test, label a 1.5 ml tube and place 250 ul of the overnight culture in the tube.
3. Take 250 ul of your phage solution and add it to each 1.5 ml tube.
4. Add 10 mM final calcium chloride to the tube. Do this by adding 5 ul of 1 M CaCl to the tube
5. Finally, add 2.5 mL of molten M17 agar (46C) and quickly mix by inverting before pouring it over the top of a M17 agar plate. You may nutate the plate in order to achieve a regular layer of agar.
6. As soon as the agar hardens, place your dish upside down and in the 37C room.
Check on your phage plaques daily and note any that form in your lab notebook. We can also take pictures of your plaques on Friday. A cool website about bacteriophage in LABs can be found here.
LAB #8: Identifying your Isolates by PCR and Sequencing
The organisms you've been working with for your cheese exhibit interesting properties. At this point, you should know what they look like, what their gram stain is, whether or not they interact with each other via growth inhibition or promotion and if they produce AI detectable by CV026. Today we will use a molecular tool to identify what genus your organism is from. We will be amplifying the 16S rRNA gene from the frozen aliquots you prepared in lab 2. This gene is well conserved among all cellular life forms - so much so that you can build a phylogenetic tree of all cellular life using an alignment of the sequences from this gene (or its homolog). To see and learn more about this tree read the primary paper by Carl Woese here.
Boiling lysis of our isolates
1. Take your 1.5 ml tubes containing your isolates in water out of the freezer and place on ice.
2. Cap each of the tubes securely with a cap lock.
3. Place them in a floating rack and add them to the boiling water. Set a timer for 5 minutes.
4. After the time is done, take your rack out of the water and place on ice immediately.
This is the lysate we will use for PCR below
PCR
We are utilizing "universal" primers to amplify the 16S rRNA gene from the isolates. Why are these primers called "universal"? Can you think of how they were designed? What organisms may they detect? Which may be excluded?
**ALL ON ICE**
For each PCR reaction you will need to generate the following mix:
| Reagent | Quantity | Calculated Total |
| Phusion 2x Mix | 10 ul | |
| 10uM F primer | 0.5 ul | |
| 10uM R primer | 0.5 ul | |
| Water | 8 ul | |
| lysate | 1 ul | ---- |
You'll be doing one PCR reaction for each of your isolates. So, for example, if I had 10 isolates, I would make up what's called a master mix. I'd need 11 total reaction (one for a "blank" that won't have DNA template added). Add 1 to this number so that you end up with enough total mix. That leaves me with 12. I'd multiply each of the amounts above by 12 to get this table:
| Reagent | Quantity | Calculated Total |
| Phusion 2x Mix | 10 ul | 120 ul |
| 10uM F primer | 0.5 ul | 6 ul |
| 10uM R primer | 0.5 ul | 6 ul |
| Water | 8 ul | 48 |
| lysate | 1 ul | ---- |
Remember to keep this all on ice! and Note that I do not multiply my lysate by 12!
1. Make your master mix following the calculated amounts above.
2. Gently mix the reaction tubes by flicking - do not pipette up and down!
3. The mix is a total of 19 ul per reaction. Label your PCR tubes according to the isolate lysate that will be added. To each tube add 19 ul of master mix. If there are bubbles in your tubes, spin them down before adding your lysate.
4. Add 1 ul of your lysate to each tube.
5. Take the tubes over to the PCR machine and we'll start the reaction. It should take about 1 hour to complete.
While the PCR machine is running, we'll take a short field trip to visit the organisms you've identified in your life list.
6. After the PCR cycle is done, take your 200 ul tubes out of the machine and place them on ice.
7. We will be transferring all 20 ul of your reactions to a 96-well plate. You need to reserve a spot on that plate by signing up on the spreadsheet located in the course conference. This way you'll know what spots are taken and when your sequences come back, we'll be able to give you your own sequences and not your classmates'.
Prof. Newton will send this plate for sequencing. Our results should come back in a few days.
Lab 9 and Final field trip: Barleycorn brewery
Today we'll be taking a trip in to Natick to visit a home-brew shop called the Barleycorn Craft Brew Shop. There we will see yeast fermentation in action. Come prepared with questions for our tour guide!
After we learn about the process of beer and soda making, we will set up our own fermentation there. It will take ~2 weeks to finish before we come back and bottle it.
Lab 9: Isolating yeast and baking bread
You might wonder how this famous eukaryotic microbe, Saccharomyces cerevisiae came to be associated with humans. The current hypothesis is that people left out their flour, which eventually promoted the growth of wild yeast. Hungry, they made bread anyway and were surprised when the dough actually rose. We've been using yeast do make our bread ever since.
Today, we'll be making the necessary containers and media to isolate our own wild yeast. Each of you will design a way to isolate yeast in a flour/water mixture we'll hand out in 200 ml flasks. By the end of the week we should see some growth in our flasks. We will use these "starters" to make bread for our final presentations.
You can find a detailed website on so-called Lambic bread cultures here.
