BISC219/F11: RNAi Lab 6

Lab 6: Series 3: Reverse Genetics- Cloning Your Gene of Interest
To see this process as an animation created by the DOLAN DNA center.
Plasmids are circular pieces of DNA that can replicate in bacteria but are not part of the bacterial chromosome. Plasmids are generally circular molecules with fewer base pairs of DNA than the chromosome and with certain sequence elements (called the origin or ori) that allow the plasmid to replicate within the bacterial cytoplasm. Many naturally occurring plasmids have been modified for the purposes of using them as research tools. For example, a gene encoding resistance to an antibiotic can be added to a plasmid so that bacteria carrying the plasmid will become antibiotic resistant. This modification allows for selection of cells that carry plasmid DNA. A simplified map of the C. elegans RNAi plasmid is below:
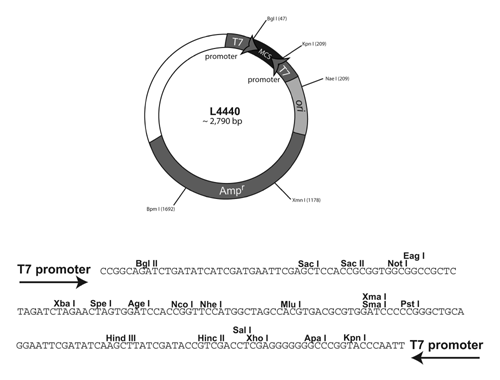
Our goal is to insert our gene of interest into the pL4440 plasmid and transform bacteria with the newly created plasmid.
Restriction enzyme digest of PCR product
To see an of this concept and process go to the Dolan DNA center website.
Once you have analyzed the agarose gel from last week and determined if the PCR amplification of your gene of interest was successful, you are ready to proceed to the next step: Restriction Enzyme Digestion of the amplified DNA.
What do restriction enzymes/endonucleases do? They are enzymes have been isolated from bacteria and cut single or double stranded DNA at specific recognition sequences. These recognition sequences are often palindromic (read the same forwards and backwards). The purpose of these enzymes in bacteria is to eliminate foreign DNA that enters the cells (ie bacteriaphage genomes) to protect the host genome. Companies now purify these enzymes from the bacteria and we can use them to manipulate DNA to link different DNA strands together.
There are two kinds of "cuts" a restriction enzyme can make, either blunt ended or overhangs called "sticky" ends. The blunt ended cuts cause the DNA to have no single stranded "overhangs" that can facilitate base pairing with another strand of complimentary DNA. This makes joining two pieces together harder, but it can be done. The "sticky" ends make joining two pieces together with complimentary base pair overhangs much easier to do and it is much more likely to happen during a ligation reaction.
For more information on restriction enzymes you can read the information on Wikipedia or my favorite site for restriction endonuclease information New England Biolabs. At NEB you can click on the different enzymes and look at the information they have available about the recognition sequence for cutting, the conditions for effectiveness and lots more.
Protocol for RE digest of bli-1
- Obtain your frozen PCR product from last week's C. elegans gene amplification from your instructor.
- Pipette 10 μL of your PCR into a clean 1.5 ml microfuge tube.
- Add 10 μL of Restriction Digest Master Mix that contains:
- 2 μL of 10X NEB restriction buffer 2 (100 mM Tris-HCl, 500 mM NaCl, 100 mM MgCl2, 10 mM Dithiothreitol pH 7.9 @ 25°C)
- 1 μL of 100X BSA (bovine serum albumin)
- 1 μL of enzyme 1 (for bli-1 that enzyme is called BglII - 10 units/ul)
- 1 μL of enzyme 2 (for bli-1 it's SpeI - 25 units/ul)
- 5 μL of dH2O
- The total volume of your reaction should be 20 μL
Incubate the reaction at 37°C for 45 minutes to allow for proper digestion.
Your instructor will have already cut and purified the pL4440 vector for you. You will need the vector for the ligation reaction. The vector can be purchased from Thermoscientific. The following website will give you more information about pL4440 [1].
Purification of samples (remove enzymes)
In order for the ligation of your new construct to be successful you MUST remove the restriction enzymes from the reaction. There are a few ways of doing this. One is to denature the enzymes with heat, although not all enzymes can be "heat killed". Thus the better option is to purify the DNA away from the enzymes (proteins) and there are wonderful kits out there to do this in a few easy steps. We will use the QIAquick PCR Purification Kit from Qiagen.
We will follow the manufacturer's instructions:
- Obtain a collection tube and spin column (contains a silica matrix) from your instructor.
- Make sure the spin column is seated properly in the collection tube.
- Add 250 μL of buffer PB to the spin column.
- Add your entire 20 μL restriction enzyme digest sample to the spin column. Close the lid tightly
- Invert a few times to mix.
- Centrifuge your sample for 1 minute at 13,200g (highest speed in your microcentrifuge).
- Separate the spin column from the collection tube; Discard the flow through in the collection tube; put your spin column back into the emptied collection tube.
- Add 700 ul of PE (Wash Buffer contains EtOH) to the spin column.
- Centrifuge for 1 minute at 13,200g (highest speed).
- Discard the flow through in the collection tube; put the spin column back into the emptied collection tube.
- Centrifuge for 1 minute at 13,200g again. This centrifugation removes any residual wash buffer that might contaminate your final elution.
- Discard the collection tube(throw it into your autoclave bag) but DO NOT discard the spin column!
- Put the spin column into a clean 1.5 ml microcentrifuge tube.
- Add 25 μL of Elution buffer to the center of the silica matrix in the spin column without touching the matrix.
- Centrifuge for 1.5 minutes at highest speed to elute your purified DNA into the microfuge tube.
- Discard the spin column in your autoclave bag. Label your elutate with your initials and the name of the C. elegans gene. This DNA will be your "insert" in the next ligation step.
- Store it in your ice bucket
Ligation into the pL4440 vector
To see an animation of this process go to the Dolan DNA center at | http://www.dnalc.org/view/15541-DNA-ligase-joining-two-lengths-of-DNA-at-their-sticky-ends.html
You have now "cut" the ends of your PCR product to make them amenable to being joined or ligated back together with other DNA molecules cut with the same restriction enzyme to make complementary base pairing possible. This is a very common practice in molecular biology to "clone" or insert genes or pieces of genes into plasmids. We will be using an enzyme called T4 DNA ligase from bacteriophage T4. For more information about ligases see Wikipedia or our exact enzyme from NEB.
Ligation Protocol:
For an effective ligation you want an excess of stick end inserts - typically the smaller piece of DNA to the plasmid, the larger piece of DNA.
We will do a 20 μL reaction.
Add 10 μL of a ligation master mix to a 0.5ml pcr tube in your team color. The ligation master mix contains:
- 2 μL of 10X T4 DNA Ligase Reaction Buffer 500 mM Tris-HCl, 100 mM MgCl2, 10 mM ATP, 100 mM Dithiothreitol pH 7.5 @ 25°C
- 3 μL of pL4440 vector that your instructor digested
- 5 μL of dH2O
To that you will add:
9 μL of insert (stored in your ice bucket after digestion and purification).
Bring your final mixture to your instructor for addition of:
1 μL of T4 DNA ligase (1000 units/ul)
Incubate the reaction at room temperature for 30 minutes.
Proceed to the transformation when the incubation is complete.
Transformation into competent cloning cells
Here are two animations from the Dolan DNA center that describe the history and the process of interspecies incorporation and expression of DNA (transformation): | http://www.dnalc.org/resources/animations/transformation1.html AND | http://www.dnalc.org/resources/animations/transformation2.html
During “transformation,” a single plasmid enters a single bacterium and, once inside, replicates and expresses the genes it encodes. In this case, the relevant genes expressed are for ampicillin resistance and for the piece of the C. elegans gene of interest. The transformation mixes were given a short time to express these gene products and then were spread on an agar plate that contained nutrients and the antibiotic ampicillin (encoded by the plasmid). Only the cells that incorporated the plasmid DNA and expressed the plasmid genes grew to form colonies of bacteria in the presence of ampicillin. The untransformed bacteria failed to form visible colonies on the ampicillin containing agar surface.
Most bacteria do not usually exist in a “transformation ready” state, but the bacteria can be made permeable to the plasmid DNA by exposing them to calcium chloride. Cells that have been treated with calcium chloride or are otherwise capable of transformation are referred to as “competent.” Competent cells are extremely fragile and must be handled gently, i.e. kept cold, not vortexed, etc. The transformation procedure is efficient enough for most lab purposes; with efficiencies as high as 107 transformed cells per microgram of DNA, but it is important to realize that only 1 cell in about 10,000 is successfully transformed.
This is especially true for "cloning competent" bacteria. The newly ligated plasmids are few in number compared to the number that can be isolated during a miniprep (you will do this more efficient isolation in a future step) so the cells are made "ultra-competent" and usually purchased from a company for A LOT of money - around $300 for 20 transformations! The newly ligated plasmid DNA is not as tightly wound as one isolated from a cell so the plasmids themselves are fragile and can be sheared and rendered untransformable. Please be gentle with your cells and your newly formed plasmids!
Transformation of newly ligated plasmid DNA into Invitrogen TOP10 cells
The Invitrogen TOP10 Chemically Competent bacterial cells are on the instructor’s bench You will transform some of your plasmid DNA into this strain. The cells are very fragile, so treat them gently. Thaw them in your ice bucket.
- Label the top or the side of the tube the cells came in with TOP10, pL4440+your gene name, and your initials or team color.
- Add 10 μL of your ligated plasmid DNA to the 50 ul of cells in the tube. Swirl gently to mix the DNA and the cells.
- Close the cap and let the transformation mixture sit on ice for 30 minutes.
- Heat shock by incubating the transformation mix in the heat block at 42°C for 60 seconds, exactly. This step must be timed exactly!!! Remove the tubes at the end of 60 seconds directly to your ice bucket for about 2 minutes.
- Pour 3 mls of Luria-Bertoni broth (LB) from the stock bottle into a clean and sterile 15ml conical tube. By doing this, you will minimize the amount of LB that will be contaminated if you accidentally touch the media with something that is not sterile. Contaminated media looks cloudy, so be sure to swirl and examine the stock bottle of LB to make sure it is not contaminated.
- Add 250 microliters of the LB in your conical tube to the transformation mix. When pipetting the media, remember to release your thumb on your micropipet slowly, to avoid splashing the liquid on the end of the barrel. The barrel is not sterile and if you see the liquid touch it, then discard the media in the waste beaker and try again with a new tip.
- Once you have added the media, close the cap and invert the tube once or twice to mix the contents.
- Incubate at 37°C for 30 minutes.
- While the plasmid DNA is being taken up by the competent cells and the new genes provided by the plasmid are being expressed by the bacteria, label two LB + amp agar plates. Label the bottom of the plates with the bacterial strain name(TOP10), the plasmid used (pL4440+bli-1), the date, your initials and team color. Label one 200 μL. You must label the bottom of the plate since the tops are easily switched.
- Put these plates in the hood with the blower on and with the lid ajar to dry the surface of the agar for about 10 minutes or until the surface looks dry but is not badly dehydrated.
- Once the transformation mix has incubated at 37°C for ~30 minutes, invert it to mix the contents and pipet 200 μL of transformed cells onto the center of one plate.
- Pipet 50 μL of transformed cells onto the second plate.
- Pour 5-10 sterile glass beads (found in a flask at your bench) onto the plates.
- Put the lid back on and gently swirl the beads all over the plates to spread the transformed bacteria around. When you are done - pour the beads into the disinfectant beaker on the instructor's bench. Do not discard them!
- Leave the agar plates undisturbed for a few minutes.
- Once they have dried enough that the surface doesn’t appear wet, invert the plates and incubate at 37°C on the shelf labeled with your lab day. Incubate for 24-48 hours. The plates should be incubated with the agar side up so that condensation will not drip onto the surface of the agar and smear the colonies that will be growing there. The 37°C incubator is by the door to the lab.
- Save the remaining transformation mix for 24 hours or until we are sure that there is at least one colony growing on each of your plates.Give it to your instructor to refrigerate for you.
What would it mean if you had no colonies on your plate? Normally, you would expect to have around 10-100 pale color colonies on each plate. After a 24-48 hour growth period the plate to allow formation of medium size but well isolated colonies, the plate should be stored in the rack in the refrigerator labeled with your lab day. If you have no colonies or a lawn of growth rather than isolated colonies, please notify your instructor right away.
Before leaving lab today, give the rest of your ligated plasmid DNA to your instructor in a labeled microfuge tube. Make sure your tube is labeled with your name, lab day, plasmid name and color coded with a piece of tape in your team color.
