BIOL478/S17:Microarray Data Analysis
Laboratory 10: Microarray Data Analysis
Wednesday, April 19, Monday, April 24, and Wednesday, April 26
Before We Begin
Viewing File Extensions
- The Windows 7 operating systems defaults to hiding file extensions. To turn them back on, do the following:
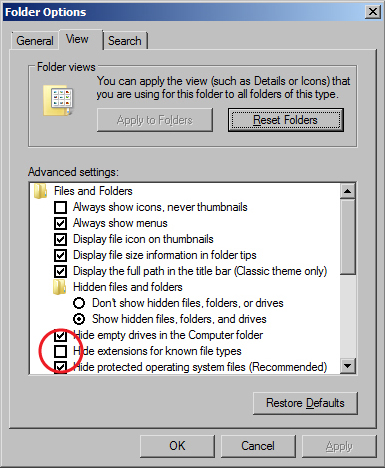
Folder Options window - Go to the Start menu and select "Control Panel".
- In the window that appears, search for "Folder Options" in the search field in the upper right hand corner.
- Click on "Folder Options" in the main window.
- When the Folder Options window appears, click on the View tab.
- Uncheck the box for "Hide extensions for known file types".
- Click the OK button.

Set Your Browser to Prompt You for the Location to Save your Downloaded Files
- In Mozilla Firefox, open the Options window.
- Select the radio button that says "Always ask me where to save files".
- You could also change the default "Save files to" location to your Desktop, so that will be the first choice when it prompts you where to save the file. (You will have to temporarily deselect the radio button to do this and then reselect it when you are done.
- Click OK to save your changes.
- In Google Chrome, open the Settings window.
- Click on the link at the bottom of the page that says "Advanced Settings".
- Check the box that says "Ask where to save each file before downloading".
- You could also change the default Download location to your Desktop, so that will be the first choice when it prompts you where to save the file.
- Your settings are automatically saved.
Background
This is a list of steps required to analyze DNA microarray data.
- Quantitate the fluorescence signal in each spot
- Calculate the ratio of red/green fluorescence
- Log2 transform the ratios
- Steps 1-3 are performed by the GenePix Pro software
- Normalize the ratios on each microarray slide
- Normalize the ratios for a set of slides in an experiment
- Steps 4-5 are performed by using a script written in the R statistical programming language and have been done for you
- Perform statistical analysis on the ratios
- Compare individual genes with known data
- Steps 6-7 are performed in Microsoft Excel
- Pattern finding algorithms (clustering)
- We will use software called STEM for clustering
- Mapping onto biological pathways
- We will use STEM for mapping the clusters onto Gene Ontology categories
- Identifying regulatory transcription factors responsible for observed changes in gene expression
GenePix Pro Protocol (Steps 1-3)
- Follow this link to download the raw image files. Put them on the Desktop.
- Right-click on the zipped file and select the menu item 7-zip > Extract here. This will extract the raw image files so that we can open them in GenePix Pro.
- Download the gene array list (GAL) file "SC4001_Yeast_MI_ReadyArray_GAL_file.gal", and put it on the Desktop.
- To download this file, right-click on the link and select "Save link as..." from the context menu that appears.
Gridding and Generating Intensity Data
- Launch GenPix Pro 7 (select Analysis Only)
- On the right hand side of the screen, select the File icon
- Select File > Open Images
- Navigate to the folder containing .tif images for your chip.
- Hold down the Control key while clicking to select both the 532 and 635 wavelength files
- Click on the Open button
- Use the brightness and contrast sliders on the upper left hand side to adjust your image to see all of the spots
- You can use the magnifying glass icon to zoom in on an area of the image, and the magnifying glass return icon to go back to the whole chip image
- Zoom out so that you can see the entire microarray slide on your screen
- On the right hand side of the screen, select the File icon
- Select Load Array List
- Select the file SC4001_Yeast_MI_ReadyArray_GAL_file.gal that you previously saved on your Desktop.
- Click the Open button
- Using the icon that has an arrow and a square, drag the grids so that they are approximately aligned with the top block of spots
- Click on the icon that looks like a compass, or a circle with cross-hairs, and select Find Array, Find All blocks, Align Features
- Alternately, you can do this in three steps by:
- Click on the icon that looks like a compass, or a circle with cross-hairs, and select Find Array
- Click on the icon again, and select Find All Blocks
- Click on the icon again, and select Align Features in All Blocks
- Alternately, you can do this in three steps by:
- Click on the icon that looks like a compass, or a circle with cross-hairs, and select Find Array, Find All blocks, Align Features
- On the right hand side of the screen, select the icon that looks like a table of data (it says "DATA" at the top of it)
- At this point, you would click on the File icon on the right hand side and select, Save Results As, and save your results as type "GenePix Pro Files (*.gpr)", along with saving a JPEG image containing all analyzed features. However, since the Department laptops do not have the license dongle, the software won't save the results. I have saved them for you and posted them on Brightspace instead.
Generating an Array Quality Report
- Launch GenePix Pro 7 (select Analysis Only)
- On the right hand side of the screen, select the File icon
- File > Open Results
- Select the desired file
- Select Report tab at the top of the screen
- On the left side of the screen, under Navigate, select the back arrow or home icon
- Select Array Quality Control
- Adjust Vital Statistic Thresholds values to:
Quality Control Limits Median signal to background > 2.5 Mean of Median background < 500 Median Signal to Noise > 4 Median % >B + 1 StdDev > 90 Feature variation < 0.5 Background Variation < 1.2 Features with Saturated pixels < 3.3% Not Found < 18% Bad Features < 7%
- Select Start
- Select Show Printable Version
- File > Print
- Under Select Printer > Select Adobe PDF
- Select Print
- Save on the Desktop
- Name the file: ChipBarcode#_yyyymmdd
- The PDF file will either open on its own or you need to open it from the file you saved it to
Looking at the raw data
- We will look at the raw data for two genes of interest, OPI1 (which should have been deleted in this strain), and NSR1, which is a gene known to be induced by cold shock.
- Open the GenePix results file (.gpr) for your chip.
- To find the data for NSR1, first go to the NSR1 page at the Saccharomyces Gene Database to learn what the systematic name (ID) for NSR1 is.
- Then search for this ID in your Excel spreadsheet
- This will tell you the block, row, and column where the NSR1 spot occurs and you can look for it in GenePix Pro.
- Repeat with OPI1 (OPI1 page at the Saccharomyces Gene Database)
- We are expecting that NSR1 should be expressed and that OPI1 should not because it has been deleted from the yeast genome. Is this the case?
Statistical Analysis (Step 6)
Downloading the Data
- Since we do not have replicate data for the Δopi1 strain, we will instead analyze wild type data previously gathered in the Dahlquist Lab.
- You have been sent an invitation to access a folder called "BIOL478 Spring 2017" on Box. From there, download the file "BIOL478_Spring2017_master_microarray_data_wt.xlsx" onto your Desktop.
- Immediately make a copy of the file with a different name which contains your name or initials to distinguish it from the master file and others in the class.
Modified t test for each timepoint
We will perform a modified t test to determine if any genes had a gene expression change that was significantly different than zero at each timepoint.
- Insert a new worksheet into your Excel workbook and name it "wt_ttest".
- Go back to the "Master_Sheet_wt" worksheet. Copy the entire sheet and paste it into your new worksheet.
- Go to the empty columns to the right on your worksheet. Create new column headings in the top cells to label the average log fold changes that you will compute. Name them with the pattern wt_<AvgLogFC>_<tx> where you use the appropriate text within the <> and where x is the time. For example, "wt_AvgLogFC_t15".
- Compute the average log fold change for the replicates for each timepoint by typing the equation:
=AVERAGE(range of cells in the row for that timepoint)
into the second cell below the column heading. For example, your equation might read
=AVERAGE(C2:F2)
Copy this equation and paste it into the rest of the column.
- Create the equation for the rest of the timepoints and paste it into their respective columns. Note that you can save yourself some time by completing the first equation for all of the averages and then copy and paste all the columns at once.
- Go to the empty columns to the right on your worksheet. Create new column headings in the top cells to label the T statistic that you will compute. Name them with the pattern <wt>_<Tstat>_<tx> where you use the appropriate text within the <> and where x is the time. For example, "wt_Tstat_t15". You will now compute a T statistic that tells you whether the normalized average log fold change is significantly different than 0 (no change in expression). Enter the equation into the second cell below the column heading:
=AVERAGE(range of cells)/(STDEV(range of cells)/SQRT(number of replicates))
For example, your equation might read:
=AVERAGE(C2:F2)/(STDEV(C2:F2)/SQRT(4))
(NOTE: in this case the number of replicates is 4. Be careful that you are using the correct number of parentheses.) Copy the equation and paste it into all rows in that column. Create the equation for the rest of the timepoints and paste it into their respective columns. Note that you can save yourself some time by completing the first equation for all of the T statistics and then copy and paste all the columns at once.
- Go to the empty columns to the right on your worksheet. Create new column headings in the top cells to label the P value that you will compute. Name them with the pattern <wt>_<Pval>_<tx> where you use the appropriate text within the <> and where x is the time. For example, "wt_Pval_t15". In the cell below the label, enter the equation:
=TDIST(ABS(cell containing T statistic),degrees of freedom,2)
For example, your equation might read:
=TDIST(ABS(AE2),3,2)
The number of degrees of freedom is the number of replicates minus one. Copy the equation and paste it into all rows in that column.
- Now we will perform adjustments to the p value to correct for the multiple testing problem.
Bonferroni Correction
- Now we will perform adjustments to the p value to correct for the multiple testing problem. Label the columns to the right with the label, wt_Bonferroni-Pval_tx (do this twice in a row).
- Type the equation
=<(STRAIN)_Pval_tx>*6189, Upon completion of this single computation, use the trick to copy the formula throughout the column. - Replace any corrected p value that is greater than 1 by the number 1 by typing the following formula into the first cell below the second wt_Bonferroni-Pval_tx header:
=IF([cell with p value]>1,1,[cell with p value]). Use the trick to copy the formula throughout the column.
Benjamini & Hochberg Correction
- Insert a new worksheet named "wt_ttest_B-H". You will need to perform the procedure below for the p values for each timepoint. Do them individually one at a time to avoid confusion.
- Copy and paste the "MasterIndex", "ID", and "Standard Name" columns from your previous worksheet into the first two columns of the new worksheet.
- For the following, use Paste special > Paste values. Copy your unadjusted p values from the first timepoint from your ttest worksheet and paste it into Column D.
- Select all of columns A, B, C, and D. Sort by ascending values on Column D. Click the sort button from A to Z on the toolbar, in the window that appears, sort by column D, smallest to largest.
- Type the header "Rank" in cell E1. We will create a series of numbers in ascending order from 1 to 6189 in this column. This is the p value rank, smallest to largest. Type "1" into cell E2 and "2" into cell E3. Select both cells E2 and E3. Double-click on the plus sign on the lower right-hand corner of your selection to fill the column with a series of numbers from 1 to 6189.
- Now you can calculate the Benjamini and Hochberg p value correction. Type (STRAIN)_B-H_Pval_tx in cell F1. Type the following formula in cell F2:
=(D2*6189)/E2and press enter. Copy that equation to the entire column. - Type "STRAIN_B-H_Pval_tx" into cell G1.
- Type the following formula into cell G2:
=IF(F2>1,1,F2)and press enter. Copy that equation to the entire column. - Select columns A through G. Now sort them by your MasterIndex in Column A in ascending order.
- Copy column G and use Paste special > Paste values to paste it into the next column on the right of your ttest sheet.
- Save your file to your flash drive and Box. Give Dr. Dahlquist access to the file.
Sanity Check: Number of genes significantly changed
- We will also perform the "sanity check" as follows:
- Go to your wt_ttest worksheet.
- Select row 1 (the row with your column headers) and select the menu item Data > Filter > Autofilter (The funnel icon on the Data tab). Little drop-down arrows should appear at the top of each column. This will enable us to filter the data according to criteria we set.
- Click on the drop-down arrow for the unadjusted p value for t60. Set a criterion that will filter your data so that the p value has to be less than 0.05.
- How many genes have p < 0.05? and what is the percentage (out of 6189)?
- How many genes have p < 0.01? and what is the percentage (out of 6189)?
- How many genes have p < 0.001? and what is the percentage (out of 6189)?
- How many genes have p < 0.0001? and what is the percentage (out of 6189)?
- Determine how many genes have a p value < 0.05 at each of the other timepoints (t15, t30, t90, t120).
- When we use a p value cut-off of p < 0.05, what we are saying is that you would have seen a gene expression change that deviates this far from zero by chance less than 5% of the time.
- We have just performed 6189 hypothesis tests. Another way to state what we are seeing with p < 0.05 is that we would expect to see this a gene expression change for at least one of the timepoints by chance in about 5% of our tests, or 309 times. Since we have more than 309 genes that pass this cut off, we know that some genes are significantly changed. However, we don't know which ones. To apply a more stringent criterion to our p values, we performed the Bonferroni and Benjamini and Hochberg corrections to these unadjusted p values. The Bonferroni correction is very stringent. The Benjamini-Hochberg correction is less stringent. To see this relationship, filter your data to determine the following:
- How many genes are p < 0.05 for the Bonferroni-corrected p value at each timepoint? and what is the percentage (out of 6189)?
- How many genes are p < 0.05 for the Benjamini and Hochberg-corrected p value at each timepoint? and what is the percentage (out of 6189)?
- In summary, the p value cut-off should not be thought of as some magical number at which data becomes "significant". Instead, it is a moveable confidence level. If we want to be very confident of our data, use a small p value cut-off. If we are OK with being less confident about a gene expression change and want to include more genes in our analysis, we can use a larger p value cut-off.
- There is one last thing to do: keeping the "Pval" filter at p < 0.05, How many have an average log fold change of > 0.25 and p < 0.05 at each timepoint? How many have an average log fold change of < -0.25 and p < 0.05 at each timepoint? (These log fold change cut-offs represent about a 20% fold change in expression.)
- Use slide 2 of this sample PowerPoint slide to see how your table should be formatted.
Comparing results with known data
- The expression of the gene NSR1 (ID: YGR159C)is known to be induced by cold shock. Find NSR1 in your dataset. What is its Log fold change at each of the timepoints in the experiment and what is its unadjusted, Bonferroni-corrected, and B-H-corrected p values? What is its average Log fold change at each of the timepoints in the experiment?
Clustering and GO Term Enrichment with stem (steps 7-8)
- Prepare your microarray data file for loading into STEM.
- Insert a new worksheet into your Excel workbook, and name it "wt_stem".
- Select all of the data from your "wt_ttest" worksheet and Paste special > paste values into your "wt_stem" worksheet.
- Your leftmost column should have the column header "MasterIndex". Rename this column to "SPOT". Column B should be named "ID". Rename this column to "Gene Symbol". Delete the column named "StandardName".
- You are now going to filter the data in such a way so that we can remove the "least significant" genes from the clustering analysis (if we keep them, the program will just be clustering noise.) Filter the data on the B-H corrected p value for t15 to be > 0.05 (that's greater than in this case). Then filter the B-H corrected p value for t30, t60, t90, and t120 to be > 0.05, without removing the filter from t15. This will select all genes that did not have a significant change in expression any any timepoint in the experiment.
- Once the data has been filtered, select all of the rows (except for your header row) and delete the rows by right-clicking and choosing "Delete Row" from the context menu. Undo the filter. This ensures that we will cluster only the genes with a "significant" change in expression and not the noise.
- Back in the "wt_stem" worksheet, delete all of the data columns EXCEPT for the Average Log Fold change columns for each timepoint (for example, wt_AvgLogFC_t15, etc.).
- Rename the data columns with just the time and units (for example, 15m, 30m, etc.).
- Save your work. Then use Save As to save this the wt_stem worksheet as Text (Tab-delimited) (*.txt). Include "wt" and "stem" in the filename to distinguish it from your other file. Click OK to the warnings.
- Note that we are going to run stem twice, once on the wt data and once on the Δopi1 data (later).
- Now download and extract the STEM software. Click here to go to the STEM web site.
- Click on the download link, register, and download the
stem.zipfile to your Desktop. - Unzip the file. You can right click on the file icon and select the menu item 7-zip > Extract Here.
- This will create a folder called
stem. Inside the folder, double-click on thestem.jarto launch the STEM program.
- Click on the download link, register, and download the
- Running STEM
- In section 1 (Expression Data Info) of the the main STEM interface window, click on the Browse... button to navigate to and select your file. We will do the wild type data first and then come back and do the Δopi1 data later.
- Click on the radio button No normalization/add 0.
- Check the box next to Spot IDs included in the data file.
- In section 2 (Gene Info) of the main STEM interface window, select Saccharomyces cerevisiae (SGD), from the drop-down menu for Gene Annotation Source. Select No cross references, from the Cross Reference Source drop-down menu. Select No Gene Locations from the Gene Location Source drop-down menu.
- In section 3 (Options) of the main STEM interface window, make sure that the Clustering Method says "STEM Clustering Method" and do not change the defaults for Maximum Number of Model Profiles or Maximum Unit Change in Model Profiles between Time Points.
- In section 4 (Execute) click on the yellow Execute button to run STEM.
- In section 1 (Expression Data Info) of the the main STEM interface window, click on the Browse... button to navigate to and select your file. We will do the wild type data first and then come back and do the Δopi1 data later.
- Viewing and Saving STEM Results
- A new window will open called "All STEM Profiles (1)". Each box corresponds to a model expression profile. Colored profiles have a statistically significant number of genes assigned; they are arranged in order from most to least significant p value. Profiles with the same color belong to the same cluster of profiles. The number in each box is simply an ID number for the profile.
- Click on the button that says "Interface Options...". At the bottom of the Interface Options window that appears below where it says "X-axis scale should be:", click on the radio button that says "Based on real time". Then close the Interface Options window.
- Take a screenshot of this window (on a PC, simultaneously press the
AltandPrintScreenbuttons to save the view in the active window to the clipboard) and paste it into a PowerPoint presentation to save your figures.
- Click on each of the SIGNIFICANT profiles (the colored ones) to open a window showing a more detailed plot containing all of the genes in that profile.
- Take a screenshot of each of the individual profile windows and save the images in your PowerPoint presentation.
- At the bottom of each profile window, there are two yellow buttons "Profile Gene Table" and "Profile GO Table". For each of the profiles, click on the "Profile Gene Table" button to see the list of genes belonging to the profile. In the window that appears, click on the "Save Table" button and save the file to your desktop. Make your filename descriptive of the contents, e.g. "wt_profile#_genelist.txt", where you replace the number symbol with the actual profile number.
- Save these files to your flash drive (or other storage). (It will be easier to zip all the files together and store them as one file).
- For each of the significant profiles, click on the "Profile GO Table" to see the list of Gene Ontology terms belonging to the profile. In the window that appears, click on the "Save Table" button and save the file to your desktop. Make your filename descriptive of the contents, e.g. "wt_profile#_GOlist.txt", where you use "wt", "dGLN3", etc. to indicate the dataset and where you replace the number symbol with the actual profile number. At this point you have saved all of the primary data from the STEM software and it's time to interpret the results!
- Save these files to your flash drive (or other storage). (It will be easier to zip all the files together and store them as one file).
- Repeat this for the Δopi1 data.
- A new window will open called "All STEM Profiles (1)". Each box corresponds to a model expression profile. Colored profiles have a statistically significant number of genes assigned; they are arranged in order from most to least significant p value. Profiles with the same color belong to the same cluster of profiles. The number in each box is simply an ID number for the profile.
- Analyzing and Interpreting STEM Results
- Which profiles were shared by the two strains and which ones were different?
- Each person in the class will select one profile for further analysis; either profile #45 or profile #22, which were the same for the two strains. Answer the following for both the wild type and Δopi1 data:
- Why did you select this profile? In other words, why was it interesting to you?
- How many genes belong to this profile for each of the strains?
- How many genes were expected to belong to this profile for each of the strains?
- What is the p value for the enrichment of genes in this profile for each of the strains? Bear in mind that we just finished computing p values to determine whether each individual gene had a significant change in gene expression at each time point. This p value determines whether the number of genes that show this particular expression profile across the time points is significantly more than expected.
- Open the GO list file you saved for this profile in Excel. This list shows all of the Gene Ontology terms that are associated with genes that fit this profile. Select the third row and then choose from the menu Data > Filter > Autofilter. Filter on the "p-value" column to show only GO terms that have a p value of < 0.05. How many GO terms are associated with this profile at p < 0.05? The GO list also has a column called "Corrected p-value". This correction is needed because the software has performed thousands of significance tests. Filter on the "Corrected p-value" column to show only GO terms that have a corrected p value of < 0.05. How many GO terms are associated with this profile with a corrected p value < 0.05 for each of the strains?
- Select 10 Gene Ontology terms from your filtered list (either p < 0.05 or corrected p < 0.05).
- You can either:
- Choose the same 10 terms that are in common between strains (if you can find any).
- Choose 10 terms that are different between the strains (5 or so from each).
- Choose some that are the same (if you can find any) and some that are different.
- Look up the definitions for each of the terms at http://geneontology.org. For your final lab report, you will discuss the biological interpretation of these GO terms. In other words, why does the cell react to cold shock by changing the expression of genes associated with these GO terms? Also, what does this have to do with Opi1 being deleted?
- To easily look up the definitions, go to http://geneontology.org.
- Copy and paste the GO ID (e.g. GO:0044848) into the search field at the upper left of the page called "Search GO Data".
- In the results page, click on the button that says "Link to detailed information about <term>, in this case "biological phase"".
- The definition will be on the next results page, e.g. here.
- You can either:
