20.109(S08):Testing cell viability (Day3)

Introduction
Promoting appropriate cell life and death is a key part of tisssue engineering. When cells are put into contact with a biomaterial (or into any novel culture condition), their viability may be affected. Some materials are cytotoxic, i.e., deadly to cells. Often, cytotoxicity varies with the concentration of one or more of the chemical components (such as a cross-linker) comprising the biomaterial, and is more or less severe for different cell types. Cell density within a culture is another factor affecting cell livelihood, notably when the number of cells exceeds the nutrient concentrations available in the culture medium. In a 3D culture such as an alginate bead, sufficient nutrients and even oxygen may not be able to diffuse to the center of the bead prior to depletion by cells on the outer rim, even when at a high concentration in the bulk fluid. Finally, note that most cells require certain soluble and/or contact-dependent signals to remain viable. For example, immune cells called naïve T cells require the cytokine IL-7 and contact with self-MHC proteins for survival.

Many assays are available to monitor the numbers of live and dead cells in a culture. The kit you will use today is made by Molecular Probes, a company (now partnered with Invitrogen) that makes a plethora of fluorescent cell stains for various purposes. The principle exploited by the LIVE/DEAD® kit is the relative permeability of cell membranes when the cell is live (intact membrane) or dead (damaged membrane). Ethidium is a nucleic acid stain that you are familiar with from running agarose gels in module 1; the ethidium homodimer-2 variant emits red fluorescence, and cannot diffuse past intact cell membranes. The dye SYTO 10, on the other hand, is membrane-permeant, and thus enters both live and dead cells; it emits fluorescence in the green channel. SYTO 10 has lower affinity for nucleic acids than does ethidium, and thus is excluded from dead cells over time, enabling one to distinguish between live (green) and dead (red) cells. Viability can be inferred by monitoring parameters other than cell permeability. For example, some membrane-permeable dyes are only activated to a fluorescent form inside cells that have active esterase enzymes, thus indicating their metabolic activity. Assays that measure cell potentials or redox activity are also available. In general, fluorescence assays are more sensitive than colorimetric assays. Along with sensitivity, stability, toxicity, and ease of scale-up are important factors to consider when choosing an assay.
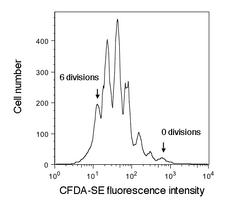
Cell vitality (or lack thereof) tells only one part of a cell culture’s story. For example, kits like the one we are using today cannot determine whether the cells assayed have divided or not. However, other dyes are available that specifically test for cell proliferation, or even distinguish cells based on what part of the cell cycle they are presently in. Proliferation assays are important for drug development, cancer research, and in tissue engineering. Total nucleic acid content is sometimes used as a measure of proliferation – Hoechst is a popular dye for this purpose. Active proliferation can be monitored by addition of 5-bromo-2'-deoxyuridine (BrdU) to cell cultures. BrdU will be incorporated only in recently synthesized DNA (S-phase cells), and can be assessed by antibody-detection after a time lag. For tracking multiple cell divisions, long-lived fluorescent dyes such as the fluorescein derivative CFDA-SE are used: about 6-10 divisions can be seen by flow cytometry (see figure at right).
Remember that cell death is just as important as cell life, and that the type of death also matters. Cells that die due to acute trauma or other tissue damage typically die by necrosis: they swell and finally burst, releasing their contents and often promoting inflammation. Under other circumstances, particularly in development and immunity, many cells undergo a programmed death called apoptosis. Unlike the more disruptive necrotic cells, apoptotic cells condense and then fragment, finally releasing membrane-contained cell bodies. Apoptosis gone awry is implicated in many diseases, and thus researchers are very interested in tracking apoptotic cells in various culture systems. Special dyes can be used to track nuclear fragmentation and other changes in early and late apoptotic cells.
Your objective today is to determine the viabilities of your three different cell cultures, and to gain experience with fluorescence assays. You are likely to encounter fluorescence and other microscopy techniques in many fields of biological engineering research.
Protocols
Today you can stagger your arrivals to lab (see today’s “talk” page). Only one group at a time will be able to work on the microscope, and assuming that cell culture setup takes ~ 1.5 hours, you will each have ~25 minutes to spend on the microscope. Please be respectful of your labmates’ time. Reading the protocol in advance will help you work more quickly, and is strongly recommended.
Part 1: Cell preparation and counting by Trypan exclusion
You will test one of your two replicates for each of your three cell samples. The cells in monolayer culture will be removed from the culture dish using trypsin (as we used for the MES cells in Module 1), while the cells in alginate must be isolated with an EDTA-citrate buffer. Otherwise, the cells can basically be treated the same way. You and your partner may want to work in parallel, one on 2D prep and one on 3D prep. Unless otherwise stated, all manipulations should be done with sterile technique.
2D culture prep
- Aspirate the culture medium from each of your samples.
- Rinse the cells with 6 mL of warm PBS, then aspirate the buffer.
- Add 1 mL of trypsin/EDTA, and incubate at 37 °C for 3 min.
- Now recover your cells:
- Add 4.5 mL of warm complete culture medium, pipet up and down, and transfer to a 15 mL conical tube.
- Spin the cells down at 1900g for 6 min (using the centrifuge that is in the TC room). Time this spin with your partner. During the spin, you might observe what your remaining replicate of the 2D sample looks like - what is the cell morphology and density like?
- Resuspend in ~5 mL of culture medium for the monolayer. You can adjust the volume a bit depending on the size of the pellet you see, but write down what you use.
3D culture prep
- Using a sterile spatula, transfer the beads from your bottom-well replicates into the next well over (to the right). This is to exclude any cells that are growing on the bottom of the plate (as opposed to actually in the beads) from analysis.
- Aspirate the culture medium from each of your samples. Be careful not to suck up the beads with the aspirating pipet, by using a serological pipet just as you did when washing your freshly synthesized beads.
- Rinse the cells with 4 mL of warm PBS, then aspirate the buffer.
- Add 3 mL of EDTA-citrate buffer, and incubate at 37 °C for 3 min.
- Now recover your cells:
- Add 3 mL of warm complete culture medium, pipet up and down to break up the beads (you may find this easier with a 1 mL pipetman rather than a serological pipet), and transfer to a 15 mL conical tube.
- Spin the cells down at 1900g for 6 min (using the centrifuge that is in the TC room). Time this spin with your partner. During the spin, you might observe what your remaining replicates of the 3D samples look like - what is the cell morphology and density like in each?
- Resuspend in ~5-10 mL of culture medium for the beads. (For high density beads, 10 mL tends to be good.) You can adjust the volume a bit depending on the size of the pellet you see, but write down what you use.
3D optional prep
- In a small Petri dish, set aside 2-3 beads for whole-construct staining if you wish.
Cell count for 2D and 3D samples
- Take 90 μL of each cell sample you want to measure into an eppendorf tube.
- Take the cell count samples out of the hood, and mix each one with 10 μL of Trypan blue solution. This is a toxic material, so please be careful not to spill it.
- Count each sample on a hemacytometer – averaging just two 4x4 squares (or counting only one if you're pressed for time) is fine. Toss your eppendorfs in the beaker labeled Cell Collect - they will be disposed of as both biological and hazardous waste by the teaching faculty.
- After you count the cells, transfer ~ 105-106 cells to a labeled eppendorf tube (for each sample). These will be used for performing the fluorescence assay. Once you have set up the first spin pf these eppendorf tubes in Part 2, you should come back to the TC room and aspirate your remaining cells, then throw away the conical tubes in the biohazard waste can by the sink.
Part 2: Preparation for Live/Dead® fluorescence assay
- Move to the main lab, and spin down your cells at 400 g for 6 minutes in a microfuge.
- Completely aspirate the culture medium from the cells, as it can interfere with staining.
- Resuspend the cells in 200 μL of dye solution (obtained from the teaching faculty).
- The dyes may be mutagenic, and should be handled with care. Wear nitrile gloves.
- Cover the tube of cells with aluminum foil, and let it sit for 15 minutes.
- Centrifuge the cell solution once more (400 g, 6 min), then take it to the fume hood.
- Remove the entire supernatant with a pipet, and expel it in the conical tube labeled Dye Collection. The dye waste will be disposed of by the teaching faculty. You should also throw the pipet tip in the solid waste container in the fume hood.
- Resuspend the cells in 50 μL of HBSS buffer.
- Mix in 950 μL of 4% glutaraldehyde solution with the cells, and incubate for another 15 minutes.
- Meanwhile, prepare for microscopy. This includes placing your memory card in the camera.
Optional: whole construct staining
- Within the Petri dish, cut your whole beads in half using a spatula.
- Rinse them with 3 mL of warm HBSS.
- Aspirate the HBSS, and pipet 200 μL of dye solution right on the beads.
- Incubate for 15 min. with the TC hood light off.
- Remove the entire supernatant with a pipet, and expel it in the conical tube labeled Dye Collection. The dye waste will be disposed of by the teaching faculty. You should also throw the pipet tip in the solid waste container in the fume hood.
- Rinse the cells with 3 mL HBSS buffer again. Aspirate off as much liquid as possible.
- Soak in 3 mL of 4% glutaraldehyde solution for 15 minutes.
- For observation, place the half-bead flat side up on a glass slide and then cover with a coverslip.
Part 3: Microscopy
When observing your cells under fluorescence excitation, you should work with the room lights off for best results. You can turn on the working lamp at the microscope bench as you set up your samples, and otherwise when you need to see what you are doing.
- If you are the first group using the microscope, you will have to turn on the microscope and allow it to warm up for 15-20 min. On the mercury lamp that is next to the microscope, first flip the ‘POWER’ switch. Next, hold the ‘Ignition’ button for about a second, then release. The lamp ready and power indicators should both be lit up now – talk to the teaching faculty if this is not the case.
- Pipet ~10 μL of your first cell sample (start with an alginate sample) onto a glass slide, then cover the solution with a round coverslip.
- Place the slide on the microscope, coverslip-side up, by pulling the left side of the metal sample holder.
- Begin your observations with the 10X objective.
- Turn on the illumination using the button at the bottom left of the microscope body (on the right-hand side is a light intensity slider).
- Next, turn the excitation light slider at the top of the microscope to ‘DIA-ILL’ (position 4).
- Try to focus your sample. However, be aware that the contrast is not great for your cells, and you might not be able to focus unless you find a piece of debris. Whether or not you find focus, after a minute or two, switch over to fluorescence. Your cells will be easier to find this way.
- First, turn the white light illumination off.
- Next, move the excitation slider to ‘FITC’ (position 3). You should see a blue light coming from the bottom part of the microscope.
- Finally, you must slide the light shield (labeled ‘SHUTTER’) to the right to unblock it. Now you can look in the microscope, and use the coarse focus to find your cells (which should be a bright green colour), then the fine focus to get a clearer view.
- You can also switch the excitation slider over to ‘EthD-1’ (position 2) to see the red-stained cells. Some of your cells may appear to be dimly red, but the dead ones are usually obviously/brightly stained.
- Be aware that the dyes do fade upon prolonged exposure to the excitation light, so don’t stay in one place too long, and when you are not actively looking in the microscope, slide the light shield back into place.
- You can try looking at your cells with the 40X objective as well if you have time. As you move between objectives and samples, choose a few representative fields to take pictures of. As a minimal data set, try to get 1-2 fields at 10X of both of your alginate samples. Post two well-captioned pictures to the wiki before leaving, so we can discuss the class data in our next lecture.
- To take a picture, remove one eyepiece from the microscope, and replace it with the camera adaptor. Be sure to keep the light shield in place until you are ready to take the picture (to avoid photobleaching)!
- Set the camera sensitivity to 800 for best results, unless your cells are really bright.
Part 4: Research idea discussion
Find a place (across the hall, in a coffeeshop, etc.) to discuss the five research results you found with your lab partner, guided by the instructions below.
Writing a research proposal requires that you identify an interesting topic, spend lots of time learning about it, and then design some clever experiments to advance the field. It also requires that you articulate your ideas so any reader is convinced of your expertise, your creativity and the significance of your findings, should you have the opportunity to carry out the experiments you’ve proposed. To begin you must identify your research question. This may be the hardest part and the most fun. Fortunately you started by finding a handful of topics to share with your lab partner. Today you should discuss and evaluate the topics you’ve gathered. Consider them based on:
- your interest in the topic
- the availability of good background information
- your likelihood of successfully advancing current understanding
- the possibility of advancing foundational technologies or finding practical applications
- if your proposal could be carried out in a reasonable amount of time and with non-infinite resources
It might be that not one of the topics you’ve identified is really suitable, in which case you should find some new ideas. It’s also possible that through discussion with your lab partner, you’ve found something new to consider. Both of these outcomes are fine but by the end of today’s lab you should have settled on a general topic or two so you can begin the next step in your proposal writing, namely background reading and critical thinking about the topic.
A few ground rules that are 20.109 specific:
- you should not propose any research question that has been the subject of your UROP or research experience outside of 20.109. This proposal must be original.
- you should keep in mind that this proposal will be presented to the class, so try to limit your scope to an idea that can be convincingly presented in a twelve minute oral presentation.
Once you and your partner have decided on a suitable research problem, it’s time to become an expert on the topic. This will mean searching the literature, talking with people, generating some ideas and critically evaluating them. To keep track of your efforts, you should start a wiki catalog on your OpenWetWare user page. How you format the page is up to you but check out the “yeast rebuild” or the “T7.2” wiki pages on OpenWetWare for examples of research ideas in process. As part of your “for next time assignment” you will have to print out your wiki page specifying your topic, your research goal and at least two helpful references that you’ve read and summarized.
For next time
1. Begin to define your research proposal by making a wiki page to collect your ideas and resources (you can do this on one page with your partner or split the effort and each turn in an individual page). Keep in mind that your presentation to the class will ultimately need:
- a brief project overview
- sufficient background information for everyone to understand your proposal
- a statement of the research problem and goals
- project details and methods
- predicted outcomes if everything goes according to plan and if nothing does
- needed resources to complete the work
You can organize your wiki page along these lines or however you feel is most helpful. For now, focus on coming up with a research problem and giving a little background about it. Print your user page(s) for next time, making sure it defines your topic, your idea and two or more references you've collected and summarized. Keep in mind that you're not committed to this idea - if you come up with something that you like better later on, that's fine.
2. Write a rough draft of your essay on tissue engineering standards (or another topic, if you clear it with me first). Next time you will give a copy of this draft to your lab partner and another copy to me, and we will both provide you with feedback by the next lab class.
