20.109(S07): Start-up signal measurement

Introduction
When we learned about M13, we saw how cleverly this virus interacts with its bacterial host, for example using a natural and common bacterial structure to gain access to the cell and then harnessing the cell’s transcription and translation machinery to make more viral particles. What we did not consider was the bacterial response to the viral attack. In fact, bacteria do “notice” viral infection and mount an SOS response within minutes. Stress response genes are up-regulated in an effort to protect the bacterial host from far more severe consequences of phage infection like lysis or destruction of its own genome.

In this next module we will turn our attention from the stress-or to the stress-ee, and consider the signals that go on inside cells to help them understand their world. It’s a complex world and so not surprisingly there exist an impressive volume and diversity of cellular messengers. Some relay information between a cell’s outer membrane and its internal processors. Messages in eukaryotic cells also travel between the nucleus and the cytoplasm, plus between and within organelles. Signaling information is transmitted through covalent modifications of the cell’s macromolecules. Common modifications include phosphate-, methyl-, and acetyl- groups, as well as peptide tags to target proteins for particular cellular locations or for degradation. The cellular signal we will consider for the next experimental module is calcium, which is not covalently added to proteins but rather modulates the shape of the proteins that bind it.
How can one study and understand a protein? For the visual folks among us, perhaps the most informative approach to understanding proteins is to elucidate their three-dimensional structures. Structures can be derived from cryoEM, NMR or X-ray diffraction patterns of the purified protein. The protein’s shape, though just a snapshot of the protein crystal, may suggest how other molecules can or can’t interact.

For example, does the structure have a narrow cleft that would restrict interactions with molecules above a certain size? Structures also reveal exquisite detail. Is a particular amino acid exposed on the surface of the protein or is it buried deep in a hydrophobic core? Does it interact with a metal or water molecule? Is the active site a shallow or deep crevice of the protein? Are there rigid portions of the structure and more flexible areas? Critically important is the idea that structural information may lead to predictions for the protein’s mechanism of action, and to testable hypotheses that examine these predictions. In lab you will have the chance to look at several proteins using a program called “Protein Explorer", examining the three-dimensional structures from all angles and identifying the location of amino acids with particular functional significance.
In this module you will study a genetically-encoded sensor, designed to measure intracellular calcium concentration. The importance of calcium for a cell will be discussed in more detail next time. Today you will become familiar with the proteins used in the calcium sensor, namely calmodulin and GFP. Today you will begin working with mouse embryonic stem cells in our cell culture facility, since these are the cells we'll use for calcium measurements.
One major objective for this experimental module is for you to learn how to perform tissue culture. Today you will learn how to get mouse embryonic stem cells to grow in a dish and also how to prevent contaminants from getting into your cell cultures. All of this will form the foundation for the ultimate experiment, which is to detect calcium signals in mammalian cells, using a fluorescent signal as a proxy.
Protocol
Part 1: Molecular modeling using Protein Explorer
The lab’s laptops should all have Protein Explorer loaded onto them. This is a free web-based viewer for biological molecules, particularly protein and nucleic acid structures. To get started type proteinexplorer.org into the Firefox browser and you’ll see the Protein Explorer’s Front Door load. Before we look at calmodulin and GFP, you should familiarize yourself with this program by examining some of the structures pre-selected in their “Atlas of Macromolecules” which can be found at the lower left portion of the Front Door. These structures are listed with their defining “PDB” identification codes. PDB, short for Protein Data Bank, is an online international resource where structural information is routinely deposited. The identification codes are four characters long, usually “#-letter-letter-letter.” Double click on the code for any protein of interest to you to get started.
- FIRSTVIEW
When the program opens, it offers you a “first view” of the structure you’ve chosen. If the "first view" does not appear you may need to follow the instructions provided by Firefox to disable popup blockers. There are three frames here to examine. On the right is the molecular image itself, rotating in space. The image includes associated water molecules, ligands and any disulfide bonds that protein might have. Try clicking and dragging on the rotating image to see what happens. The second frame is the “Control Panel” on the upper left. In this frame, you can modify the image shown in the molecular image panel (more on this feature in a minute). The control panel also has links to places for more information about the structure being examined. Finally there is the “Message Frame” on the lower left. With this panel you can specifically query the identity of any atom shown in the image panel. Try clicking on the image in the image panel and then read the message frame that results. You’ll see information about that atom’s identity. Commands can also be entered into the Message Frame, using the box just above where information is reported.
Try working for just a minute with the control panel. First use it to hide or show any water molecules (shown as red spheres in the image panel) that were associated with the solved structure. When the water molecules are hidden, what is left should be the backbone trace of the protein, with different colors indicating different backbones. Any associated ligands also remain. Use the “backbone trace” link in the control panel to explore what this term means, then click the “back” button to return. The “back” button is your escape hatch and will always link you to the standard view of the control panel. Next click on any ligand in the image view panel (if there are any) to discover the identity of these molecules. The identity will be shown in the message panel in the lower left. The final aspect of the control panel to become familiar with is the “explore more features” link. Information from the biologists who solved the structure you are examining is included here in three categories: “about”, “reliability” and “substructures.” Explore and enjoy these now.
- QUICKVIEW
Once you are finished with the FirstView study, you can move on to the “QuickView” features. With this part of the program you can modify and customize the image displayed in the image viewer. Again, you can choose any molecule from the menu to begin and click the “quick views” link in the control panel. There are three pull-down menus that are displayed: “select” “display” and “color.” Try a few things in each of these menus just to see what happens to the image that’s displayed. Don’t forget you can still rotate the image using your mouse in the image frame. With each change you make from the pull-down menus, helpful information should appear. Also keep an eye on the message panel to follow the story. Finally, there are a few buttons below the three drop-down menus. These offer quick ways to make commonly desired changes to the structure’s appearance. Give them a go.
Here’s one thing you might want to explore: how does the polarity of the amino acids differ on the surface of the protein vs in the core? The “slab” button might be particularly useful here. This is just one idea. Feel free to explore any aspects of protein structure that are of interest to you. Note, however, that Protein Explorer is not able to “dock” two protein files together or mutate particular residues in the structure you are considering.
- CUSTOMIZING
Once you’re happy with your mastery of Protein Explorer’s basic workings, use the NCBI web site to find a PDB ID for some calmodulin structures. Choose the "Structure" database from the dropdown menu and search for "apo calmodulin". You'll find several entries since calcium-modulated proteins exist in so many cell types and in complexes with many different targets. Once you’ve chosen a PDB ID (singular or plural), go back to the center panel at the bottom of the Protein Explorer front door and type this code into the box associated with “Enter any PDB identification code here.” Then “go.” After you've examined the protein structure in the absence of calcium find a structure for "ca bound calmodulin." Compare the apo and bound structures and write a few sentences in your lab notebook describing the calcuim-induced change in conformation. Note the PDB IDs of the structures you're comparing, the resolution of the 3-dimensional structure, the origin of the proteins you're examining and any other information you consider relevant and useful. The final protein structure that you should become familiar with is that of "green fluorescent protein" (aka GFP). Choose a few structures to examine, ideally from different organisms. Make some notes about similarities and differences in these structures.
Part 2: Microscopy
You learned about the lab's fluorescent microscope on the very first day of the term but that almost certainly seems like a lifetime ago and a refresher will be useful for everyone. The microscope has a white light switch on the front of the stage base and a slider to vary the light's intensity. There is also a mercury lamp to the left of the scope that you will not need today but will be essential once the cells are fluorescent.
You should examine some of the mouse embryonic stem cells that are used in Part 3 of today's lab. A sample of recently trypsinized cells resuspended in growth media will be provided. Invert the tube with cells to resuspend them and then use a drop of the liquid (7 ul) to make a slide. Add a coverslip to prevent the sample from drying out. Find the cells under the 10X objective (yellow), starting with the stage low and slowly turning the course adjustment away from you to raise the stage until you see the cells come into focus. Note that cells will move when you move the stage; if you've focused on microscope dust the things you're seeing will remain steady even as the stage moves.
Next you should move the 40X objective (blue) into place and use only the fine adjustment to refocus the cells. Using the microscope this way should prevent broken coverslips and scratched lenses. You and your partner should both practice finding the cells.
Once you've found a cluster of cells you like and think are representative of the slide as a whole, use the lab's digital camera to capture the data. Download the image to your laptop and include a printout in your lab notebook for today.
Part 3: Intro to cell culture
In the past century, we have learned a tremendous amount by studying the behavior of mammalian cells maintained in the laboratory. Tissue culture was originally developed about 100 years ago as a method for learning about mammalian biology. The term tissue culture was originally coined because people were doing exactly that, extracting tissue and letting it live in a dish for a short time. Today, most tissue culture experiments are done using cultured cells. Much of what we know about cancer, heritable diseases, and the effects of the environment on human health has been derived from studies of cultured cells.

What types of cells do people study, and where do they come from? Cells that come from a tissue are called primary cells, because they come directly from an animal. It is very difficult to culture primary cells, largely because primary cells that are placed in culture divide a limited number of times. This limitation in the lifespan of cultured primary cells, called the Hayflick limit, is a problem because it requires a researcher to constantly remove tissues from animals in order to complete a study. To get around this problem, people have studied cells that are immortal, which means that they can divide indefinitely.
One type of familiar immortalized cell is the cancer cell. Tumor cells continuously divide allowing cancer to invade tissues and proliferate. Cancer cells behave the same way in culture, and under the right conditions, cells can be taken from a tumor and divide indefinitely in culture. Another type of immortalized cell is the embryonic stem cell. Embryonic stem cells are derived from an early stage embryo, and these cells are completely undifferentiated and pluripotent, which means that under the right conditions, they can become any mammalian cell type. Mouse embryonic stem cells have become a valuable research tool, and it is this cell type that we will be using for this experimental module.
The art of tissue culture lies in the ability to create conditions that are similar to what a cell would experience in an animal, namely 37°C and neutral pH. Blood nourishes the cells in an animal, and blood components are used to feed cells in culture. Serum, the cell-free component of blood, contains many of the factors necessary to support the growth of cells outside the animal. Consequently, serum is frequently added to tissue culture medium, although serum-free media exist and support some types of cultured cells.
Cultured mammalian cells must grow in a germ-free environment and researchers using tissue culture must be skilled in sterile technique. Germs double very quickly relative to mammalian cells. An average mammalian cells doubles about once per day whereas a bacterium is able to double every 20 minutes under optimal conditions. Consequently, if you put 100 mammalian cells and 1 bacteria together in a dish, within 24 hours you would have ~200 unhappy mammalian cells, and about 100 million happy bacteria! Needless to say, you would not find it very useful to continue to study the behavior of your mammalian cells under these conditions!
Each of you will have a 25 cm2 flask of mouse embryonic stem (MES) cells that you will use to seed a six-well dish. You and your partner will seed the dishes at different concentrations so you should decide who will seed at 1:100 and who will seed at 1:400.
- Prewarm all the required reagents in the water bath.
- Look at your cells as you remove them from the incubator. Look first at the color and clarity of the media. Fresh media is reddish-orange in color and if the media on your cells is yellow or cloudy, it could mean that the cells are overgrown, contaminated or starved for CO2. Next look at the cells on the inverted microscope. Note their shape and arrangement in the dish and how densely the cells cover the surface.
- Move the cells into the sterile hood, as well as the PBS, trypsin, and media that you will need. One of the greatest sources for TC contamination is moving materials in and out of the hood since this disturbs the air flow that maintains the sterile environment inside the hood. Anticipate what you will need during your experiment to avoid moving your arms in and out of the hood while your cells are inside.
- Aspirate the media from the cells.
- Wash the cells by adding 5 ml PBS. Tip the flask back and forth to rinse all the cells, and then aspirate the liquid out of the dish.
- To dislodge the cells from the dish, you will add trypsin, a proteolytic enzyme. Using a 2 ml pipet, add 2 ml of trypsin to the flask. For one minute precisely (use your timer), tip the flask in each direction to distribute the trypsin over the cells then aspirate the trypsin off the cells. Incubate the cells (“dry”) at 37° for 10 minutes, again using your timer to precisely time this incubation.
- While you are waiting, you can add 1 ml of gelatin to each well of a six-well dish. This should be done in the sterile hood with sterile technique. The gelatin will be removed before you seed the dish with your MES cells but it is important to pre-treat the dish this way. The gelatin must remain in the wells for at least 10 minutes.
- With a 5 ml pipet, add 6 ml of media to the trypsinized MES cells and pipet the liquid up and down (“triterate”) to remove the cells from the plastic and suspend them in the liquid. Remove a small amount of the liquid to an eppendorf tube and take it to the inverted microscopes.
- Fill one chamber of a hemocytometer with 10 ul of the cell suspension. This slide has an etched grid of nine large squares. The square in the center is further etched into 25 squares each with a volume of 0.1 ul and 16 tiny chambers (4x4 pattern). The concentration of cells in a sample can be determined by counting the cells that fall within the 4x4 pattern and then multiplying by 10,000 to determine the number of cells/ml. You should count the cells in the four corner squares of the 25 square grid, then average the numbers to determine the concentration of cells in your suspension.
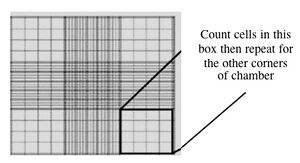
Counting cells using a hemocytometer - You and your partner will seed at different concentrations. Decide if you will try the 1:100 or 1:400 dilution and add the appropriate amount of cell suspension to 10 ml of media in a 15 ml conical tube.
- Remove the gelatin from the six-well dish you have prepared and add 3 ml of your cell dilution to each well. Be sure to label your dish with your name, today’s date, the cell line (called “J1”) and the type of media you have used. Return your cells to the incubator.
- Aspirate any remaining cell suspensions to destroy them and clean up the hood. Dispose any vessels that held cells in the Biohazard waste and any sharps in the grey bins. The next group who uses your hood should find the surfaces wiped down, no equipment left inside, the sash closed and the germicidal UV lamp on.

DONE!
For next time
- Familiarize yourself with the calcium-induced conformational changes of calmodulin by reading the Zhang et al 1995 paper in Nature Structural Biology We can provide you with a copy if you don't can't access this article through the MIT library.
- Calculate the number of cells in each well of your six-well dish. Determine how many cells there should be next time if:
- only 25% of the cells are able to stick and proliferate (this is called a 25% plating efficiency),
- the doubling time for the cells is 24 hours, and
- the cells take 24 hours to recover from trypsin treatment before they begin doubling.
- Review your understanding of protein structure visualization by examining another protein we'll be working with this term. Search the NCBI structural database for a variant of the green fluorescent protein that is mutated so the Valine at position 68 is a Leucine. Identify where the mutated position is in the 3D structure and then print out an image of the protein structure with the mutation clearly indicated (in pen is fine). If you're having trouble getting this program to run or print or anything, just do this part of the assignment when you get to lab!
- In anticipation of the calcium assays we will perform next time, please calculate the amount of CaCl2 (fw 110.99) needed to make 50 ml of a 20 mM solution.
Reagents list
- Trypsin
- 0.25% Trypsin
- 1 mM EDTA
- D-PBS
- Dulbecco’s Phosphate-Buffered Saline
- J1 ES Cell Culture Medium
- 100 U/ml Penicillin/Streptomycin
- 0.3 mg/ml Glutamine
- 0.1 mM BME
- 1 mM Non-Essential Amino Acid (NNEA)
- 10% Fetal Bovine Serum (Atlantic Biologic, Inc., Atlanta, GA)
- Leukemia Inhibitory Factor (LIF) - LIF helps stem cells maintain their undifferentiated state
- Gelatin
- 0.1% TC-grade gelatin prepared in H2O
- Gelatin is a protein prepared by partial hydrolysis of collagen
