30 nm chromatin fiber, from Robinson et al PNAS 2006
In the upcoming experiment we'll see how nature has overcome a particularly challenging design issue, namely space constraints in the nucleus. A cell's dimensions do not increase linearly with DNA content. Instead, eukaryotic cells remain compartmentalized and cells compact their DNA by wrapping it around assemblies of histone proteins called nucleosomes. Nucleosomes wrap around eachother to form chromatin. This packaging of the DNA solves the space issue, allowing a meter or so of DNA to be crammed into a space perhaps 10 um across, but creates a new problem. Wrapped DNA is less accessible to the transcription and replication machinery and so chromatin becomes a barrier to reactions that a cell must undergo to survive. Nature's answer to this chromatin barrier: multiprotein complexes that redistribute nucleosomes to make active genes accessible. It remains unclear if the redistribution of nucleosomes is a cause or a consequence of every gene's activity but one thing that's exquisitely clear is that chromatin remodeling is required for appropriate gene expression which is, in turn, required for healthy cell behaviors.
We'll study one chromatin-remodeling complex called SAGA in this experimental module. A recent structure for the complex was elucidated through electron microscopy Wu et al 2004. The yeast SAGA structure, shown in the figure below, can be imagined to "dock" with the DNA and associated proteins, allowing us to propose some very elegant models for how chromatin-modifications might be performed and regulated. Though yeasts are separated from humans by 1.6 billion years, their SAGA complexes are biochemically similar. Indeed, complexes like the S. cerevisiae SAGA complex are found in many evolutionarily distant eukaryotic cells. What's even more remarkable is that these SAGA complexes have identical numbers of protein subunits and the proteins have notable sequence homologies, suggesting conserved functions even in cells with diverse life-styles like yeast and human cells. Thus, there is good reason to believe that an understanding of how SAGA works in yeast can give us insight into its role in cells more medically relevant, like human cells. And yeast are a lot easier to genetically and biochemically manipulate than human cells. For example, in the first few days of this module, we'll add a protein tag to some SAGA genes in yeast or some sequences that are regulated by SAGA. If we wanted to genetically manipulate humans this way, it wouldn't be so simple, either experimentally or ethically.
a model for SAGA's interaction with transcription factors and nucleosome-bound DNA, image courtesy of P. Schultz
The name "SAGA" is an acronym for "Spt-Ada-Gcn5-acetyltransferase." A combination of biochemical and genetic data suggested that the GCN5 gene (the G in SAGA) encoded an enzymatic activity, namely a histone-acetyl transferase, that exists as a large protein complex. The authors named this complex "SAGA." Grant et al 1997. Nineteen proteins, including GCN5, associate to form SAGA. A table of these subunits, and some genomic information is tabulated here.
DNA is compacted by wrapping it around nucleosomes to form chromatin
chromatin structure must be modified to enable appropriate gene expression, replication and repair of the DNA,
large protein complexes are responsible for modifying chromatin structure,
SAGA is one example of a chromatin modifying complex,
the overall structure of SAGA is conserved from yeast to humans
SAGA seems to have the same 19 proteins in many kinds of cells.
Given these facts, you may be surprised to learn that not all the SAGA proteins are absolutely required for yeast cells to live. It's possible to delete the gene for a nonessential subunit from the yeast genome and the cell can still grow and divide, although sometimes with impaired functions. We'll exploit the fact that some SAGA genes are not essential for viability in this experiment.
With this series of experiments we'll modify a non-essential SAGA subunit or a gene that's regulated by SAGA, and then consider the consequence of this modification on gene expression. The kind of modification we'll make (namely addition of a "TAP tag") is generally considered neutral in terms of cellular function. But the point of departure for this module is to test that assumption. Is it really safe to assume that a tagged protein complex works in the same way in a cell as the non-tagged version does? Today you will design some primers to add the TAP-tag to a SAGA or SAGA-regulated gene of your choosing. Later in this experimental module, you will examine your tagged strain for changes in gene expression, looking for new phenotypes associated with tagged strain as well as looking by DNA microarray for genes whose expression is affected by the modification of this subunit. By the end of this module you'll likely know more than anyone in the world about how these modifications affect yeast gene expression, and you'll convey your findings in a research article suitable for publication.
Protocols
Part 1: Choosing a gene to TAP tag
You have two options for which gene you'll study this module. Option 1: You can study one of the lesser known, nonessential subunits of SAGA.
Option 2: you can study one of the "unknown open reading frames" that seems to be affected by deletion of a SAGA subunit, namely sgf73.
Gene expression details are included in the table. A positive number in the "log 2(green/red)" column indicates more expression of unknown open reading frame when SGF73 is present than when its absent. A negative number indicates more expression of the unknown ORF when SGF73 is deleted from the strain. If there are two numbers in the log2 column, then you have the data for two gene expression comparisons. The "green signal" and "red signal" columns express relative expression rates in arbitrary units. Again, if there are two numbers, then there are two measurements that have been made.
Begin by acquainting yourself with the genes you're interested in. You can find relevant information in the Saccharomyces Genome Database for the subunits. You can also search Pubmed to find out a little more about the role of the gene you've chosen in gene expression. You might also look for homologs of your gene by pasting the gene name into P-POD, the Princeton Protein Orthology Database, or look for interactions and pathways relevant to that protein by pasting the gene name into bioPIXIE.
Do not shortchange yourself on this part of the experiment, since you will be working with the gene you choose today for the rest of the module.
NOTE: The nomenclature for S. cerevisiae is precise and helpful. Wild type genes are normally given an italicized, three letter acronym based on the phenotype of a mutation in that gene. So a HIS gene is unable to make histidine if the gene is defective (of course, dead cells all have only one phenotype so this presumes loss of the gene product doesn't kill the cell...). Since there exist several genes that can give rise to similar phenotypes, related genes are given a number as well, e.g. HIS3, HIS4, etc. To describe recessive mutant alleles, lower case letters are used. So a strain that is his3 has a mutation that affects the function of the HIS3 gene. Since there can be several different mutations described for any given gene, a second number gets associated with the mutant, e.g. his3-1 or his3delta200. Proteins are distinguished from DNA by capitalizing only the first letter of the gene product: the HIS3 gene makes the His3 protein. Naturally there are exceptions to these rules, but in general you can pretty confidently follow them.
Part 2: Designing your TAP-tagging oligos
Everyone's starting strain is called NY411, which has the following genotype:
This genotype tells you that the strain is haploid and of the "A" mating type. The genotype also tells you that the strain cannot make its own histidine, lysine, leucine, uracil, or tryptophan due to the indicated mutations in the HIS4, LYS2, LEU2, URA3 and TRP1 genes. These mutations have insignificant effects when the strain is grown on rich or "complete" media but no growth occurs when one of those needed media components is left out, or "dropped out" as such media is usually described.
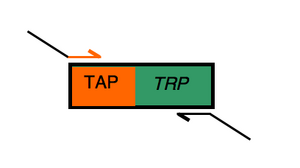
design of TAP-tagging oligos
Everyone will modify the gene of their choosing by adding a "TAP tag" (details of the "TAP tag" will come on Day 4 of this module) that's linked to a TRP1 gene. Successful modification of the gene with the TAP-TRP sequence will restore growth of the strain on "SC-trp," which is media lacking tryptophan ("SC" for "synthetic complete;" "minus trp" for the absence of tryptophan).
The primers you design today will have two parts. One part of each primer will be sequences identical to the TAP-TRP fusion, enabling the primers to anneal to the TAP-TRP gene fusion that we have on a plasmid, and amplify it by a PCR that you will perform at the end of lab today.
The second part of everyone's primers will be "tails" or "flaps" that will allow the amplified TAP-TRP sequence to modify the C-terminus of the gene of interest. These "flap" sequences must be at least 39 bases long to allow sufficient specificity and recombination frequency once the amplified fragment is transformed into yeast cells. The total length for the primer you're designing will be 59 nucleotides since oligonucleotide synthesis companies change their pricing structure and recommendations for oligos 60 bases and longer. This leaves you 20 bases for the "landing" sequence that will annealing to the TAP-TRP fusion during PCR. Bottom line: limitations in the synthesis technology impose the 59 base limit, but fortunately this turns out to be minimally intrusive for experiments like the one you'll start today.
Designing the "forward" primer
You and your partner should begin by opening a new MSWord document to create a "primer record" for the sequences you are designing. Put your names at the top, your team color, today's date and a short description of what you are trying to do.
Begin your primer design by noting the "universal" landing sequence that will be used to anneal to the start of the TAP-TRP fusion:
tcc atg gaa aag aga aga tg
landing sequence for forward primer Paste this sequence into the IDT oligo analyzer tool to determine (and note on your primer record!) the Tm. Recall that the first few rounds of PCR must be performed at a temperature below the melting temperature of this landing sequence (5° below is the rule of thumb) if these 20 bases are to bind the template DNA. Later you will add 39 base "flaps" to the primers which will still be present during the PCR but during the first rounds of PCR, they will have no complementary sequences to which they can anneal. Thus the reactions must start below the melting temperature ("Tm") of the landing sequence.
Next you must add the primer "flap." Begin this process by retrieving the genomic DNA sequence for the gene you've chosen as it's listed at SGD. You will need 39 bases from the 3' end of the gene (NOT INCLUDING THE STOP CODON!!) to serve as the "flap" for your forward primer. Copy these 39 bases to your MSWord document. This is the sequence that will recombine with the 3' end of the gene you have decided to modify.
Paste these 39 bases to the 20 TAP bases in your forward primer. Will you paste the 39 bases to the left (i.e. "upstream" or "5'") or to the right (i.e. "downstream" or "3'") of the TAP landing sequence? If you are unsure, please ask one of the teaching faculty.
Distinguish the landing sequence from the flap by making one sequence uppercase and the other lower case. Alternatively underline or italicize one section. Note how they are distinguished on the MSWord document you have started as the primer's record.
Use the OligoAnalyzer from Integrated DNA Technologies to find the Tm and GC content for the full forward primer you've designed. Note these values on the primer record.
Great. Now you're ready to design the second of the primer pair. Many of the same steps are involved but it's a little trickier since you will have find the complementary sequence to the ones listed by SGD, and you'll have to reverse the primer at the very end so it reads in the conventional 5'to 3' direction.
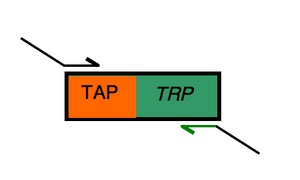
Designing the "reverse" primer
Note that this protocol for designing the reverse primer is just one method of many that can work. For example, taking the reverse complement can be done at any stage, making an identical primer. So if these steps don't seem sensible to you, try a way that does.
Begin by noting the 20 bases of "landing" sequence that will anneal to the TRP gene on our PCR template:
tac gac tca cta tag ggc ga
These are written in the 5' to 3' direction already for the reverse complement. landing sequence for reverse primer. You should determine the Tm of this sequence using the OligoAnalyzer program from IDT.
Next find the flap sequence for the reverse primer by finding the 39 bases that follow the stop codon of the gene you'll study from SGD. The custom retrieval feature is useful for this: find the "retrieve sequences" bar on the right hand side of the page you're working on, choose "custom retrieval" from the menu and then "view." Finally, add 39 bases downstream in the custom retrieval box that's on the bottom left of the page. Don't choose reverse complement if you plan to follow these instructions precisely, since you'll take the reverse complement of just the 39 base pair flap in the next step. You can look here at a table of the genetic code if you're unsure of the stop codon sequence.
Choose the "GCG" format for the DNA region and select the 39 bases that follow the stop codon (do not include the stop) to serve as your "flap" sequence.
Use OligoAnalyzer to take the reverse complement of the flap. Be sure to delete the numbers that paste in with the genetic code since the OligoAnalyzer program is expecting only G, A, T or C in your sequence.
Add the flap sequence (now in its reverse complement form) to the landing sequence. Think carefully about which end of the landing sequence you should paste it to. If you have questions or are uncertain here, please ask. Distinguish the landing sequence from the tail in the same way you did for the forward primer. Note which end is the 5' end of the reverse primer.
Last thing is to use OligoAnalyzer to find the Tm, GC content etc for reverse primer. Does it matter if you're looking at the top strand sequence or its complement?
Paste the sequences for the primers you've designed into a table of SAGA-related information on your wiki userpage. You should also print out copies of the primer record for your lab notebooks and one copy for your team to hand in.
The last thing to do is to compare the sequence of the primer pair you've designed to the ones we have pre-ordered for the class. These are the ones that are available for you to use for PCR today and they are listed among the reagents list at the end of today's lab.
Part 3: PCR
Before you begin this portion of the lab, it is a great idea to wash the barrels of your pipetmen with a paper towel and 70% EtOH. You could also wash your bench area.
All the components necessary for performing PCR are available from the teaching faculty, including primers like the ones you just designed. Your reactions will contain the following:
Template + or - 3 ul pBS1479 (= 1 ug)
Forward Primer 2 ul (=200 pmol)
Reverse Primer 2 ul (=200 pmol)
PCR Master Mix* 40 ul of 2.5X stock (see REAGENTS LIST)
"Mg Solution" 2 ul of 25mM stock
H2O to final volume of 100 ul
The PCR Master Mix contains buffer, dNTPs and Taq Polymerase
You will assemble three PCR tubes, two complete reactions and another without template. The "no template" reaction serves as a control for contamination.
Begin by getting 3 PCR tubes from the teaching faculty and adding the correct amount of water to each. The volume you need with template should be added to two tubes and that amount +3 ul should be added to the third PCR tube.
Next add the primers to each reaction. Be sure to change tips between additions.
Next add template to two of the three reaction tubes.
Finally add PCR Master Mix and also the Magnesium Solution to each tube, pipetting up and down to mix. Leave your tubes on ice until the entire class is ready to load reactions into the thermal cycler.
The reactions will undergo the following PCR cycle:
94° 5 minutes
94° 30 seconds
48° 30 seconds
72° 4 minutes
repeat steps 2-4 34 more times
72° 10 minutes
4° forever (or until one of the teaching faculty removes the reactions and stores them in the freezer)
DONE!
For next time
Your major assignment for this experimental module will be a formal research article describing your work. Some general requirements for this report are detailed on the class wiki. Start by (re)reading these guidelines. You'll write part of the introduction today, first reading the relevant primary literature, and then writing three paragraphs according to the suggested scheme below. This scheme is just a rough framework to help you organize your thoughts. Naturally you are free to apply your personal style and writing approach. One thing everyone must do: keep track of the sources for your information to properly reference them in your final paper.
Paragraph 1: most general of all. You don't have to start with the dawn of creation or how the first cell came to exist but you might consider framing the experiments around some larger questions like:
why is gene expression important?
how do nucleosome positions relate to gene expression?
what relevant modifications of nucleosomes have been described?
what tells nucleosomes where to bind?
what tells nucleosomes when and where to move?
Paragraph 2: introduction of SAGA as a chromatin remodeling complex. This paragraph can't possibly cover all that's known about SAGA but some relevant and interesting aspects you might address are the:
distribution of the complex: is S. cerevisiae the only SAGA-containing cell on the planet? is SAGA found at every gene in S. cerevisiae?
biochemistry of the complex: number of subunits, how these were identified, are they all necessary for SAGA stability? for SAGA structure? do they form subcomplexes? are there shared subunits with other chromatin remodelers?
genetics of the complex: what happens when you delete subunits? what about pairwise deletions? are there traditional phenotypes associated with SAGA mutations? are there disease states associated with mutations in any of the subunits in organisms more complex than yeast?
structure of the complex: how was this determined? are there other structural views that are supportive or contradictory? does the structure support any genetic or biochemical data?
genes regulated by the complex: has SAGA been associated with every gene? with particular transcription factors? with particular cellular responses? how were such experiments performed? are there supportive or contradictory studies?
Paragraph 3: introduction to the gene you'll be modifying. You chose that gene to tag for some reason; here's the chance to say why. In addition to your personal interest in the subunit you should provide some fundamental information from SGD about the gene and protein, like:
chromosomal location
protein size
protein features (acidic patches, structural or sequence motifs, etc)
phenotypes associated with deletion of the gene (if any are known)
synthetic phenotypes associated with deletion of gene in presence of other mutations (if any are known)
interaction data from experiments like two-hybrid or GST-pull downs?
You and your lab partner can and should discuss the papers you find and you should help each other understand them. You can also ask the teaching faculty if you are unclear on the details of some technique you read about. When it comes time to write, you must do so on your own. You and your lab partner will hand in individual assignments. Please submit this part of the assignment electronically to both nkuldell and astachow AT mit DOT edu. Good luck and have fun!
Reagents list
PCR Master Mix (2.5X)
62.5 U/ml Taq DNA Polymerase
125 mM KCl
75 mM Tris-HCl, pH 8.3
3.75 mM Mg(OAc)2
500 uM each dNTP
Std PCR reactions (50 ul final volume)
~100 ng template
~100 pmole each primer
1X concentration of all reagents in 2.5X mix
denature 94-95°C
anneal 5°C less than lowest primer hyb temp
extend 1’/kb to be amplified
Mg Solution
provided with the PCR mix
labelled only as "Mg2+" so it's not clear what the anion ion is
TAP-Tagging Primers
primer name
primer number
sequence
SGF73_TAP-TRP_fwd
FO6043
5'ATTGGAAATTCTGTGAACCCCTACAATGGCAGAATAAAT tcc atg gaa aag aga aga tg
SGF73_TAP-TRP_rev
FO6044
5'CTC ACT TCG TGA ACA TGC TGG ATA ACG TGC ATG ATT CAA tac gac tca cta tag ggc ga
SGF29_TAP-TRP_fwd
NO236
5'CTGCCTTCGCCAACGGCTTTGGCAAACCTAGCAAGGAAA tcc atg gaa aag aga aga tg
SGF29_TAP-TRP_rev
NO237
5'GAA GAT CTT ATG ATA TGT AGT AAA TGT TAA CCA CCA TTG tac gac tca cta tag ggc ga
SGF11_TAP-TRP_fwd
NO238
5' GCTCATTTACAGAGATGTTTGAGTAGGGGTGCTAGACGT tcc atg gaa aag aga aga tg
SGF11_TAP-TRP_rev
NO239
5'TCT GTG CCT TTT CAA TTA CCC ATA AAC CAC CAC CTA GTG tac gac tca cta tag ggc ga
UBP8_TAP-TRP_fwd
NO240
5' CAGGCATATTTATTATTCTACACCATTCGTCAAGTAAAT tcc atg gaa aag aga aga tg
UBP8_TAP-TRP_rev
NO241
5'TTT TTG TTT TAT TAT TAT TGT TGA ATG CTA TTT GCT GAA tac gac tca cta tag ggc ga
SUS1_TAP-TRP_fwd
NO242
5' CAAATAAGGGAATTTCTTGAAGAGATTGTAGATACACAA tcc atg gaa aag aga aga tg
SUS1_TAP-TRP_rev
NO243
5'TGT AAT AAT ATT GGG AAT TAA GGT GCA TTT TCG TAT CCT tac gac tca cta tag ggc ga
Unk1_TAP-TRP_fwd
NO244
5' GTAGCACATAGAGAAAACATCGCTTTCCCTCCGCAATTT tcc atg gaa aag aga aga tg
Unk1_TAP-TRP_rev
NO245
5'TTT TTT TTT TTT TAA TCC GGT AAA AAA AAG GGA ATA TTC tac gac tca cta tag ggc ga
Unk2_TAP-TRP_fwd
NO246
5' GACTACATATCTGACCACATCTGGAAAACTAGCTCCCAC tcc atg gaa aag aga aga tg
Unk2_TAP-TRP_rev
NO247
5' GTG GTT TGT GAT AGA ATT TCT GAT TAT TAA GCA ATG AAA tac gac tca cta tag ggc ga
Unk3_TAP-TRP_fwd
NO248
5'AAGGTTAATTTTGACATCGAGGAAGAGCAAGAAGGACAA tcc atg gaa aag aga aga tg
Unk3_TAP-TRP_rev
NO249
5'AGA TTT ATC TGA TAT GCT CAA TTT CCC CTC CCA TTT TCA tac gac tca cta tag ggc ga
Unk4_TAP-TRP__fwd
NO250
5'ATGAAACAATCTAAGAAAAAAACTTCTTTCACCAGATTC tcc atg gaa aag aga aga tg
Unk4_TAP-TRP_rev
NO251
5'CAA CCT AGC TCC TAT CAA GTT CTT ATT ACC TTC ATT TTA tac gac tca cta tag ggc ga
Unk5_TAP-TRP__fwd
NO252
5'TGTTGTCTATGTTTAATTAACCTATGTTGTGACGTTTTT tcc atg gaa aag aga aga tg
Unk5_TAP-TRP_rev
NO253
5'GAT GTT CTT TCC ATA CTA ATT TTT AGC TGT CGA TTA GAA tac gac tca cta tag ggc ga
Unk6_TAP-TRP_fwd
NO254
5'CAAGAATTCTCAGCTTCTTCAACTGACAATAAACAAAGT tcc atg gaa aag aga aga tg
Unk6_TAP-TRP_rev
NO255
5'CTT TGG CCA TTA TTT TAT TTG GCT AAA AAT TTC AAT GTT tac gac tca cta tag ggc ga
Unk7_TAP-TRP_fwd
NO256
5'CGTTTTTGCATTTCCTTTCCCTGTTTTGGATTGAGTATA tcc atg gaa aag aga aga tg
Unk7_TAP-TRP_rev
NO257
5' CAT GGT ACC TGC TGG AAG AAC TTT GTT GTT TGT TTA GAT tac gac tca cta tag ggc ga
Unk8_TAP-TRP_fwd
NO 228
5' GT ATT TGT CGA TTA CAA AAC ACA TCC TGT AGG CGC AAA tcc atg gaa aag aga aga tg
pic.jpg)