Biomod/2011/Harvard/HarvarDNAnos:LabNotebook Evan
<html> <head>
<style>
- column-one { display:none; width:0px;}
.container{background-color: #f5f5f5; margin-top:50px} .OWWNBcpCurrentDateFilled {display: none;}
- content {width: 0px; margin: 0 auto auto 0; padding: 1em 1em 1em 1em; align: center;}
- column-content {width: 0px; float: left; margin: 0 0 0 0;padding: 0;}
.firstHeading {display:none; width:0px;}
- globalWrapper{width:1280px; margin:auto}
body {background: #F0F0F0 !important;}
- column-one {display:none; width:0px;background-color: #f0f0f0;}
- content{border:none;margin: 0 0 0 0; padding: 1em 1em 1em 1em; position: center; width: 800px;background-color: #f0f0f0; }
.container{ width: 800px; margin: auto; background-color: #f0f0f0; text-align:justify; font-family: helvetica, arial, sans-serif; color:#f0f0f0; margin-top:25px; }
- bodyContent{ width: 1267px; align: center; background-color: #f0f0f0;}
- column-content{width: 1280px;background-color: #f0f0f0;}
.firstHeading { display:none;width:0px;background-color: #f0f0f0;}
- header{position: center; width: 800px;background-color: #f0f0f0;}
- footer{position: center; width:1280px;}
</style>
</head> </html>
<html>
</html>
Evan's Lab Notebook
Summer
Week 1
Day 1 (2011-06-06)
Miscellaneous Notes
- 5 nm gold particles
- width of DNA helix = 2nm
- 7249 bp M13 ssDNA
- streptavadin + biotin
4 main box ideas
- middle-separating cylinder/box
- middle-separating sphere (http://www.sciencemag.org/content/332/6027/342.full.html)
- cylinder with vertical seam
- cylinder with patch
Day 2 (2011-06-07)
- completed design for middle-separating box using hexagonal cadnano
- got CanDo output for design
- met with Tom to generate ideas about boxes



Evan's middle-opening hexagonal box:
- Screenshot of CanDo fluctuations video:

- caDNAno JSON file: Media:Boxwithmiddleopening.json
Day 3 (2011-06-08)
- completed design for box with lid using square cadnano
- CanDo gives box that splits in two - likely because of inadequate staple strands
- fixed staples, CanDo now gives robust box with very flappy lid
Evan's Denmark-like boxwithlid (single hinge)
- CanDo fluctuations video: Media:Boxwithlidcando.avi
- caDNAno JSON file: Media:Boxwithlid.json
- Via Tom's idea
- Will need more than one hinge I think, but not sure how to do that in cadnano without making inner loop
Day 4 (2011-06-09)
- worked on cadnano design of 2D Han disk
- attended Yin group meeting
Day 5 (2011-06-10)
- finished design of Han's 9-layer concentric ring
- attended Shih group meeting
- realized that reversal of all strands makes it impossible to match my staple strands with the ones listed in Han's supplementary info
- began learning Maya via learning videos and a DNA tutorial
Evan's caDNAno implementation of Han et al's 9-layer concentric ring (using curved DNA): File:Nineconcentricrings.json
- all DNA strands (scaffold and staple) are in reverse orientation, making it impossible to match my staple strands with those reported by Han et al in their supplementary information.
Week 2
Day 6 (2011-06-13)
- created accurate 3D model of the Han sphere using tori in Maya
- using the radii for each ring as provided in the supplementary information
- also made table with calculations for phi and height of each ring
- worked on implementing lock mechanism for boxwithlid
- added several staple strand hangovers in cadnano (not done yet)
Day 7 (2011-06-14)
- worked on lock mechanism for boxwithlid
- decided to implement azobenzene-type mechanism, with azobenzene (removed with UV light) or other strand (with same sequence as azobenzene, removed via toehold displacement) bringing together staple strand from lid and staple strand from box
- added lock staples to cadnano file: File:Boxwithlidwithlockmechanism.json
- generated an excel file with staple sequences (actual lock area of staples to be added): File:Staple Sequences for boxwithlidwithlockmechanism.xlsx
- here is a schematic of the box, with dimensions and lock staples: File:Boxwithlid locking mechanism schematic.pdf
- Updates to schematic and additional notes:
- all staple locks have been changed to stand alone (two staple locks on same side of box or lid will not be connected inside box or lid)
- nomenclature: sub L will refer to the lid part of the lock; sub B will refer to the box part; N_L7 will refer to the staple lock in the north edge of the lid, coming out of helix 7
- the height of the box is equivalent to only 41 ss nucleotides
- the scaffold for the boxwithlid is 6084 nt
- do not forget that the lid is 2 nm thick
- sequence lengths for lock staples:
- 5-6 Ts for u-turn
- for all except N_L: 25 nt lock area, leq 35 nt inside box or lid [ box - 5 Ts (for u-turn or height of lid) - 5 Ts (for area that does not anneal to azobenzene) - 15 nt (for azobenzene annealing area) - 2 nt spacer (for azobenzene TT) ]
- for N_L: extra 5 Ts needed in lock area for lateral displacement to N_B
- azobenzene properties
- 32 nt
- F_n = 5' - CGTGATTGTTACCAG TT CGTTAGTTCAGACAG - 3' (not F_12X because positions of X's not shown)
- ssDNA persistence length = 1.4 nm
- no hairpin needed for u-turn
- no need to worry about helical turn position at beginning of lock area of lock staple
- check for secondary structures using NuPack for ssDNA coming off of box or lid
- add -TT to ends of helices on bottom of box to prevent blunt end stacking
- ideas for improving tightness of box
- add "foamy mess" of ss staples at ends of helices at top of box
- uses interactions between ss staples at ends of helices at top of box to make a "fence" around the opening
CanDo result of Evan's boxwithlid with a double hinge: Media:Boxwithliddoublehinge.avi
- Looks much better than with single hinge
- See my lab notebook entry from today for more information and source files
Day 8 (2011-06-15)
- Random notes:
- in cadnano, arrowheads refer to 3' ends, squares refer to 5' ends
- the interior of the boxwithlid has dimensions 16 nm (8 helices) x 14 nm (7 helices) x 10.88 nm (32 bp)
- finished designing azobenzene-type lock mechanism for boxwithlid, after two realizations:
- opposite lock staples (e.g. N_B and N_L), as produced yesterday, were antiparallel, but the actually need to be parallel
- lock staple couples (e.g. the two N_B's) need to be parallel, or azobenzene strand could just bind the couple together instead of binding box to lid
- hence, schematics from yesterday should be ignored with respect to azobenzene-type lock mechanism. new ones will be produced tomorrow. the schematic from yesterday is however still roughly accurate with respect to the antiparallel lock mechanism
- cadnano file for boxwithlid with azobenzene-type lock mechanism now has filename boxwithlid_parlockmechanism.json, whereas cadnano file for boxwithlid with yesterday's antiparallel opposite lock staples is now boxwithlid_antiparlockmechanism.json
- the latter could be used for a different box with different locking mechanism
- generated excel file with staple sequences, and inserted -TT ends and lock area sequences
- should be ready to order staples as soon as toehold for F_n+ is designed
Day 9 (2011-06-16)
- completed new schematic of boxwithlid with parallel lock mechanism:

- called Nanocs, a company that makes 5 nm streptavidin-coated gold nanoparticles ($320 for 1 mL), to ask them about the thickness of the streptavidin. a man with very bad english answered the phone and told me that the company estimates that the streptavidin adds about 3 nm to the diameter of the particle, but that they don't really have real data.
- worked on designing toehold for F_n+ that would not produce secondary structures (using NUPACK to check), but was largely unsuccessful
- attended biweekly BioMod meeting with Yin, Shih, Adam, and other postdocs. some ideas:
- instead of using gold nanoparticle for cargo, make small origami "nanoball," triangle, or ring structure to fold into box
- one-part folding (folding origami around cargo) is more robust than two-part folding (folding origami, then placing cargo into origami in open state, then closing origami)
- solving the problem of holes on top and bottom of sphere by making it into a torus (copying the uppermost ring down through the sphere)
- making a enclosed torus space by polymerizing DNA rings (very large diameter to height ratio would make gaps between rings small)
- decided to order strands tomorrow for most basic designs and to better synthesize ideas on group wiki (see project page)
Day 10 (2011-06-17)
- traditions of listening to Chinese version of "Friday" and taking group photo
- successfully designed toehold for F_n+ so that there are no significant secondary structures
- F_n+ toehold: 5' - ... - AGACCGGCAT - 3'
- F_n+* toehold complement: 5' - ATGCCGCTCT - ... - 3'
- analysis with NUPACK worked after realizing that it does not accept a poly-T "box" representation, where there lock areas protrude into the hairpin. so I put the poly-T in between the two lock areas instead, so it could form a hairpin with the lock areas protruding outward
- NUPACK analysis was run at 25 degrees C, and concentration of "box" was 1 nM, "lock" was 1 nM, and "key" was 1 microM
- read up on disulfide bond formation and cleavage. see protocols page for more info.
- worked with Adam on editing boxwithlid_parlockmechanism design. finished edits, and prepared staples for ordering
- will put up relevant files on data page
- next step: design box with parallel lid helices so that the jagged interface between box and lid will be complementary
Edited .json caDNAno file of boxwithlid_parlockmechanism and Excel file of staples for box using azobenzene sequence (F_n) lock:
- Media:Boxwithlid parlockmechanism-edited.json
- Media:New Staple Sequences for boxwithlidwithlockmechanism.xlsb
NUPACK analysis of the "lock" F_n+; F_n+ with the minimal "box"; and F_n+ with the "box" and the "key" F_n+*:



Week 3
Day 11 (2011-06-20)
- created schematics
- 5' - 5' disulfide linking

- 5' - 3' disulfide linking and subsequent origami folding

- started on schematic of nanoparticle chain
- pK_a of -SH is 8.3
- brief meeting with Tom
- what happens if only one side of the boxwithlid locks first? does that tilt the lid so that the opposite side can't lock?
- based on Smith, Cui, and Bustamante (1996), we think that the force of shear lock binding is about 50 pN per lock no matter the number of bps that anneal. the implications of this force for the lid can be calculated using beam bending equations that engineers use.
- Nick talked to him about his idea of using a strand on the outside of the sphere to coax the signal strand that solubilizes the cargo into the sphere - increasing effective concentration by using two-part binding
- strands for box were ordered today from IDT
Day 12 (2011-06-21)
- 5' 5' disulfide linking experiment:
- diluted "M13-barcode 1" and "U1C" strands to 200 nM, checked concentration on Nanodrop, then diluted to 100 nM
- see this excel file for calculations: File:Dilution calc.xlsx
- gold nanoparticle chain experiment:
- finished schematic of nanoparticle chain
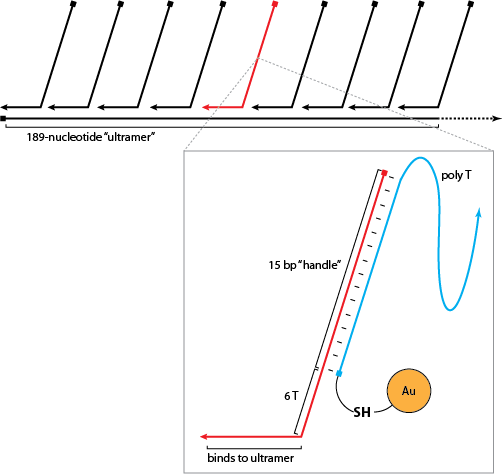
- diluted ultramer to 20 uM and staple strands to 200 uM, checked concentrations on Nanodrop, then diluted to 10 and 100 uM respectively
- created annealing reaction ladder in PCR tubes in 100 uL reaction volume with 100 nM ultramer and 1 uM staples: reaction 1 has just ultramer, reaction 2 has ultramer plus first staple, reaction 3 has ultramer plus first two staples, etc.
- placed ladder in thermal cycler, to be retrieved tomorrow
- see this excel file for calculations: File:Dilution calc.xlsx

- learned how to use DLS machine from Steve (see protocols page), how to make agarose gel to separate origami from Adam (see protocols page), and watched Adam use TEM
- to do: revise 5' 3' disulfide linking diagram (divide into two separate diagrams)
Day 13 (2011-06-22)
- ran 6% PAGE TBE gel of annealing reaction ladder for nanoparticle chain experiment, but gel was torn up before we could scan it
- ladder = 4 uL in 20 uL water
- 10 uL of each sample + 2 uL loading dye

- worked on wiki Project Outline page
- created own gold nanoparticles with Steve using the protocol "Preparing Colloidal Gold for Electron Microscopy" (Polysciences, Inc.) for 5 nm, and his own recipes for 15 nm and above
- the ones bigger than 15 nm were made by adding extra layers of gold to the 15 nm particles

- ran a 10% PAGE TBE gel of annealing reaction ladder, with successful results (see Data and Output page)
- 45 mins, 100 V (next time run for 1.5 hours so more separation)
- 2 uL ladder in 20 uL water
- it seems that the handles are annealing correctly (upward trend)
Day 14 (2011-06-23)
- worked on presentation for Yin lab meeting
- with Adam:
- added ~ 10 mg of phosphine to 50 mL aliquots of Steve's 5 nm gold particles synthesized yesterday.
- these are in 50 mL conical tubes, covered in foil (the phosphine is light sensitive) and shaking slowly on the rotary shaker (the same thing we use to stain the gels on)
- the next step (tomorrow) is to add NaCl to these nanoparticles which will change the color to blue and allow us to precipitate them, which is necessary for further steps in the conjugation protocol
- to measure AuNP concentration, use the approximate linear relationship: 1 A520 unit = 1 uM nanoparticle concentration
Day 15 (2011-06-24)
- Friday traditions of group photo and Chinese Friday
- ran two gels for nanoparticle chain experiment
- one was a rerun of the gel from Day 13 in order to get more separation
- the other only had ladder and several lanes of the reaction mixture #10, which has has the ultramer + all nine handles
- this is so we can extract the complete chain base (ultramer + nine handles)
- both gels were run at 10% TBE, 2 hours, 100 V, 1.5 uL ladder in 20 uL water, 9 uL sample in each lane
- the former gel was taken out after 2 hours and here is the gel scan
- because there was not much separation for the last sample lane, we ran the latter gel for an additional 2 hours (total of 4 hours)
- stained it with SYBR-Gold, so actually can't use this get to extract the complete chain base
- but the good news is that there is enough separation after 4 hours for us to extract the top band (which should be just ultramer + 9 handles); here is the gel scan.
- will have to make more of reaction 10 (ultramer + all nine handles) on Monday and rerun gel using a DIFFERENT STAINER
- Sherrie and Nick added thiolated oligos to the gold nanoparticles as per this protocol
- will run a gel on the result after the weekend
- worked with minimal box test today
- resuspended minbox, F_n+, and F_n+* to 100 uM
- created diluted stocks of the three strands: minbox diluted stock = 100 nM, F_n+ diluted stock = 100 nM, and F_n+* diluted stock = 1000 nM (1 uM)
- three PCR tubes:
- tube 1: 10 uL 10x folding buffer, 10 uL minbox diluted stock, 80 uL water
- tube 2: 10 uL 10x folding buffer, 10 uL minbox diluted stock, 10 uL F_n+ (lock) diluted stock, 70 uL water
- these two tubes were then put on the thermal cycler for 1 hour: heating to 70 degrees C, cooling to 25 degrees C
- then in tube 3: 50 uL of tube 2 after annealing, 1 uL fresh 10x folding buffer, 10 uL F_n+* (key) diluted stock (this step happened at 5:20pm
- all three tubes are just sitting at room temperature over the weekend; a gel will be run on Monday
Week 4
Day 16 (2011-06-27)
- Boxwithlid Pool Labels - Order #7026079
- "Box 1 P1" = Plate 1: A1-H6 = Box Core = 90 oligos
- "Box 1 P2" = Plate 2: A1-E5 = Box Bottom Staples = 53 oligos
- "Box 1 P3" = Plate 2: E6-F11 = Box Top Staples with Extensions = 18 oligos
- "Box 1 P4" = Plate 2: F12-G7 = Lid Core Staples = 8 oligos
- "Box 1 P5" = Plate 2: G8-H3 = Lid Edge Staples = 8 oligos
- NOTE: NO LOCK STAPLES IN THIS ORDER
- Folding Boxwithlid (without lock staples) for the first time
- in a PCR tube, combined for a total of 50 uL:
- 6.75 uL (# oligos * 0.075) P1
- 3.975 uL P2
- 1.35 uL P3
- 0.6 uL P4
- 0.6 uL P5
- 5 uL 10x folding buffer
- 16.7 uL water
- 15 uL M13mp18 ssDNA
- placed in middle thermal cycler Block 2B, under Wei's program "72HR"
- should be done folding at 5pm on Thursday
- in a PCR tube, combined for a total of 50 uL:
- minimal box experiment

First PAGE Gel (Ladder, Minbox, Minbox + Lock, Minbox + Lock + Key, Ladder)
- ladder spilled into adjacent lanes

Second PAGE Gel
- nanoparticle chain experiment
- made a fresh ultramer + 9 handles tube, annealed on thermal cycler
- need to run gel (WITHOUT SYBR-GOLD) tomorrow to extract the species we want
Day 17 (2011-06-28)
- nanoparticle chain experiment
- ran gel of annealed ultramer + 9 handles solution at 100 V for 4.5 hour
- tried looking at gel under UV lamp, but bands were too light to distinguish
- stained with SYBR-Gold, but bands still not visible using UV plate
- scanned gel using Typhoon - still no clear bands
- realized that the SYBR-Gold that we have been using for the past few days has gone bad - explains why all of our gels have been so light
- will redo gel again tomorrow, stain with different solution of SYBR-Gold, and extract relevant band
- helped Sherrie fold her sphere!
- added Python script to wiki
- revised schematic of disulfide bond solubilization method to reflect comments from last week's group presentation
Day 18 (2011-06-29)
- realized at beginning of day that we had been using 10% TBE-Urea Gels - Urea denatures all dsDNA so this is BAD - don't use Urea!!!
- minimal box experiment
- ran new gel, using 10% TBE (no urea) gel, for 1 hr at 100 V, stained with SYBR-Gold: here is the gel scan - it seems that you might be able to see locking/unlocking, but unclear - decided needed to do another gel with more lanes so we have more info to compare the different lanes against
- in 7 PCR tubes, ran Adam's NPRTF themal cycler program
- tube 1 (ladder): nothing (tomorrow, for gel, add 0.5 uL ladder, 20 uL water, 2 uL LD)
- tube 2 (box): 10 uL of previous reaction tube 1 (add 2 uL LD tomorrow)
- tube 3 (lock): 1 uL F_n+ diluted solution, 1 uL 10x buffer, 8 uL water (add 2 uL LD tomorrow)
- tube 4 (key): 1 uL F_n+*, 1 uL 10x buffer, 8 uL water (add 2 uL LD tomorrow)
- tube 5 (lock+key): 1 uL F_n+, 1 uL F_n+*, 1 uL 10x buffer, 7 uL water (add 2 uL LD tomorrow)
- tube 6 (box+lock): 10 uL of previous reaction tube 2 (add 2 uL LD tomorrow)
- tube 7 (box+lock+key): 1 uL box, 1 uL F_n+, 1 uL 10x buffer, 7 uL water (after annealing, added 1 uL F_n+*; tomorrow add 2 uL LD)

Third PAGE Gel (Ladder, Minbox, Minbox + Lock, Minbox + Lock + Key)
- nanoparticle chain experiment
- ran another gel of the ultramer + 9 handles solution for 4 hrs at 100 V - stained with SYBR-Safe (10 uL in 100 mL water, Bryan recommends pre-staining next time)
- ran a gel scan (there was a band we could distinguish!), and used that to estimate where to extract what we want: here is an overlay of the post-cut gel on top of the gel scan
- Ralf helped us do AFM imaging of the spheres we folded overnight
- Adam prepped the spheres for TEM
Day 19 (2011-06-30)
- Wei helped us TEM our first sphere origami
- It is unclear if the spherical objects are actually our spheres or just random dirt/water
- Need to do a better staining job next time
- Sherrie and I extracted the DNA out of the gel extract obtained yesterday from the ultramer + 9 handles gel, as per "Filtration and recovery" from this protocol
- Had biweekly BioMod meeting with Peng and William. Notes:
- Wei: .15 @ 260nm on nanodrop indicates correct concentration of origami
- disulfide linkers are not compatible with gold cargo -> we should focus on using IDT photocleavable spacers instead now
- gold will fold with 1-pot folding because of Magnesium
- consider passivating the nanoparticle with poly T, or coating with A-A*, B-B*
- or put it into a origami barrel
- M13 scaffold gel band is suspicious in Sherrie's sphere gel -> buy new M13 scaffold!
- Ran gel of revamped minimal box experiment, 1.25 hrs, 100 V
- Here is the scan

Successful minimal box experiment PAGE gel!
- shows locking and unlocking of box via strand displacement
- Started thinking about cargo loading mechanism for boxwithlid
Day 20 (2011-07-01)
- Boxwithlid
- Took first folding (mixed on Day 16) out of thermal cycler
- Ran gel of folded origami as per protocol, 80 V, 2 hours, except:
- the agarose gel had 1 mL 1 M MgCl2 and 8 uL SYBR-Gold
- M13mp18 lane had 2 uL scaffold, 8 uL water, 2 uL Bryan's loading buffer
- origami lane had 10 uL sample + 2 uL Bryan's loading buffer
- used 12 uL Bryan's 1kb ladder
- used 10 mM MgCl2, 0.5X TBE running buffer
- scanned the gel
- using UV plate, extracted the band indicated by grey box in the gel scan, and purified this extract as per the protocol
- Gave Ralf an aliquot of the purified origami to AFM
- Wei prepared a sample of the purified origami to TEM

Scan of gel with 1000 bp ladder, M13mp18 scaffold, and folded origami Boxwithlid without lock staples
- grey box indicates section of gel that was extracted for origami purification
- designed new staple strand for box bottom so we can load and solubilize gold nanoparticles using internal photocleavable spacers and U1C strand attached to nanoparticle (Au-5ThioMC6-GAACTGGAGTAGCAC 15-mer oligo)
- Staple 64[52]->67[52]: GTGCTACTCCAGTTCTTTTT/iSpPC/TTTTTAAATCCCGCGCCCAATAGCAAATCGTAGGAATCATTATACGT
Week 5
Day 21(2011-07-04)
Happy 4th of July!
Day 22 (2011-07-05)
- created new header image for the Project Outline page
- created Order #4 .xls spreadsheet with lock staples (previously not ordered) and cargo loading strand for the Boxwithlid - to be discussed with Adam and placed
- First folding of Boxwithlid (with no lock staples)
- on the evening of Day 20, Wei TEM-ed the purified Boxwithlid first folding sample but saw nothing
- today, retrieved AFM images that Ralf took, also on the evening of Day 20, of the same purified sample (different tube, though)
- there are some box like structures in these images, with possible lids, but these "lids" could also just be AFM image artifacts
- to confirm, Adam will TEM the UNPURIFIED sample tonight to see if he can see anything
- Wrote up protocol on PAGE gels




AFM images taken on 2011-07-01 by Ralf of folded origami Boxwithlid without lock staples (purified)
- looks like a box with a lid, but the "lid" could just be an scanning artifact
Day 23 (2011-07-06)
- calculated that sphere volume is 13.75 times larger than the volume of boxwithlid
- internal photocleavable spacers are very expensive (~$150/oligo), so we can't rely too much on them
- typed up protocol for using the Typhoon gel scanner
- worked on organizing, populating Tutorials page
- TEM-ed the first folding of the boxwithlid
- saw many long rectangular white spots, but the dimensions are off, so we have decided to refold the box
- Boxwithlid refold ("Box 2")
- made working stock: 90 uL P1, 53 uL P2, 18 uL P3, 8 uL P4, 8 uL P5
- 2 PCR tubes with:
- 13.275 uL working stock (0.075 * 177 oligos)
- 5 uL 10x TAE 12.5 mM folding buffer
- 15 uL M13mp18 scaffold (new, from NEB, 111 nM)
- 16.725 uL Nuclease-free water
- tube 1: Shawn Douglas' "6 Day" thermal cycler program
- tube 2: Wei's "72HR" thermal cycler program
- New sphere folding (closed and open-via-no-equator-staples, with correct buffer concentrations) imaged on the AFM and TEM today: looked promising
- Ran gel of of this new folding as well: tomorrow purify the spheres out of this gel
- Also tomorrow: AuNP-DNA Conjugation, retrieve TEM images, TEM close sphere if TEM is working, maybe TEM folded boxes again
Day 24 (2011-07-07)
- Preparing of AuNP Conjugation (following this protocol)
- Nanodrop to determine concentration of nanoparticles (UV-Vis, A520 nm wavelength, 1 mm pathlength): One tube had a reading of around 1.15, the other had a reading of around 2.01, so we decided to combine them, and got a final reading of 1.6375, corresponding to a concentration of 16.375 uM
- Nanodrop to determine concentration of the "M13" oligo (which is a 5'-thiolated U1C strand with a 3' poly T): (2933 ng/uL) * (17.9 nMoles/0.31 mg) [from spec sheet] = 169 uM
- Prepared a TEM grid of the nanoparticles so we can confirm their size tomorrow
- Based on the observed concentrations, if the size is correct, tomorrow we will combine for 40 uL total reaction:
- 3 uL M13
- 30.96 uL nanoparticles
- 2 uL 10x TBE buffer
- 0.4 uL 5M NaCl
- 3.64 uL water
- Prepared TEM grids for purified spheres and other tubes of purified first folding of box
- Typed up protocol for AFM
- AFM-ed purified closed sphere (did not see much) and one of the other tubes of purified first folding of box
Day 25 (2011-07-08)
- Typed up the protocol for the NanoDrop and tutorials for the different types of buffer in the lab and the different types of water in the lab
- TEM-ed some of the grids prepared yesterday with Wei
- We did not prepare the nanoparticle grid correctly: we DO need to glow discharge, then add 3.5 uL of the sample, let sit 4 mins, wick off excess liquid, no stain
- Did not see any closed spheres
- Did not see any boxes in left-side sample
- After doing these three samples, we caused the TEM lose its vacuum
- For origami, Wei recommends that we use the NanoDrop UV-Vis at 260 nm to check that the absorbance of the sample is between 0.1 and 0.4 - indicates that there is a high enough concentration of origami
- If not concentrated enough, Wei says we can use a spin column to remove some of the solvent
- Although we have not confirmed the size of the nanoparticles with TEM, because Nick double-checked the consistency of the DLS measurement, we decided to proceed with the AuNP-DNA conjugation
- We created two 40 uL tubes of the following, to sit on the slow shaker over the weekend:
- 3 uL M13
- 30.96 uL nanoparticles
- 2 uL 10x TBE buffer
- 0.4 uL 5M NaCl
- 3.64 uL water
- We created two 40 uL tubes of the following, to sit on the slow shaker over the weekend:
- Emailed Jean to set up time for TEM training
- Adam will order the lock staples and cargo loading staple for the box
Week 6
Day 26 (2011-07-11)
- Took out 72 hour folding of Box 2 from thermal cycler and ran gel of it
- 2.5 hours, 80 V, 2% agarose, 10 + 2 uL Bryan's LD
- can't see band distinct from the scaffold, so will wait until seeing tomorrow's 1 week folding reaction before deciding whether or not to attempt purification
- Took off of slow shaker one tube of the AuNP Conjugation reaction (68 hours)
- Ran gel of the sample alongside plain AuNPs following this Sucrose Extraction protocol - 4% agarose base, 3% agarose top layer
- Modifications: 80 V for 3 hours, 100 V continuing (Adam will stop it later tonight); each lane has 10 uL sample, 4 uL 30% sucrose
- Ran gel of the sample alongside plain AuNPs following this Sucrose Extraction protocol - 4% agarose base, 3% agarose top layer
- How to remember orientation of gel box vs. gel: "Run to red, black to black"
- Attempted AFM of unpurified 72 hour folding of Box 2 - but did not see anything

Gel Scan of 2nd Boxwithlid Folding (see lane to the very right)
- 72 hour folding only
Day 27 (2011-07-12)
- Gold Nanoparticle Chain Experiment
- Amicon purified AuNP-DNA Conjugates that were run through a gel and sucrose extracted by Adam and Ralf last night (no separation into bands occurred, not sure why)
- Nanodropping lead to conclusion that the filter did not work properly, hence we decided to proceed using the AuNP-DNA Conjugates that were not run through the gel and that were not purified
- Nanodropping for chain assembly
- AuNP-DNA Conjugates, A520 on UV-Vis: 1.275 = 12.75 uM
- Ultramer+9 handles (chain base), A260 on Nucleic Acid: 13.05 ng/uL = 0.0751 uM
- Two tubes
- 1:9 chain base:NP mole ratio = 18.864 uL chain + 1 uL NP
- 1:45 chain base: NP mole ratio (5-fold excess) = 3.77 uL chain + 1 uL NP
- Let sit at room temperature since 12:30pm
- Amicon purified AuNP-DNA Conjugates that were run through a gel and sucrose extracted by Adam and Ralf last night (no separation into bands occurred, not sure why)
- 2nd Folding of Box
- Took out 6 day folding in the morning
- Made a non-annealed "blank" sample with 13.275 uL working stock, 5 uL 10x folding buffer, 15 uL M13 scaffold, and 16.725 water; did not put in thermal cycler at all
- Ran 2% agarose gel (1 mL 1 M MgCl2, 8 uL SYBR-Gold) (80 V, 2 hours; then 100 V, 1 hour )
- Lane 1: 12 uL 1 kb ladder
- Lane 2: 1 uL M13 scaffold, 9 uL H20, 2 uL Bryan's Loading Buffer
- Lane 3: 10 uL non-annealed Box 2 , 2 uL LB
- Lane 4: 5 uL 72 hr folding, 5 uL water, 2 uL LB
- Lane 5: 5 uL 6 day folding, 5 uL water, 2 uL LB
- The thermal cycling causes some sort of aggregation, but we suspect that the origami is not folding properly, as the scaffold band is prominent in all the lanes (it should go away, because there is an excess of staples)

Gel Scan of All of 2nd Boxwithlid Folding
Day 28 (2011-07-13)
- Why is the box not folding?
- Wrong staple sequences: double checked caDNAno output sequence with what was ordered, seemed okay
- The design simply does not fold
- Bad folding buffer: made new folding buffers today, see below
- Perhaps the MgCl2 concentration is too low: the 10x Folding Buffer I've been using has had 12.5 mM of MgCl2
- Too much single-stranded DNA: Box 3 Folding includes lock staples, so should remedy this possible issue
- Boxwithlid lock staples came in today, resuspended all of them to 100 uL by adding (# nmoles * 10) uL of water to each tube
- Created Box 3 Pre-stocks: 10 uL of each oligo
Label Contents Wells Number of Staples P1 Box Core (without cargo-loading strand) All of Plate 1 (without F12) 89 P2 Box Bottom Plate 2 A1-E5 53 P3 Box Top Plate 2 E6 - F11 18 P4 Lid Core Plate 2 F12 - G7 8 P5 Lid Edge Plate 2 G8 - H3 8 P6- Cargo-loading Strand without Dock Plate 1 F12 1 P7 Lock Staples Tubes 16
- P6+ will be the Cargo-loading Strand WITH Dock
- Created Box 3 Working Stock, where each staple has concentration of 520 nM = 100 uM/193 (total number of staples)
- Made 10x Folding Buffer with Four Different MgCl2 Concentrations (2 mL each)
- 12 mM Final MgCl2 Concentration: 240 uL 1 M MgCl2, 200 uL TE, 1560 uL ddH20
- 15 mM: 300, 200, 1500
- 18 mM: 360, 200, 1440
- 21 mM: 420, 200, 1380
- Folded Box 3, with Wei's Protocol, using three types of 10x Folding Buffer: 15 mM MgCl2, 18 mM MgCl2, and 21 mM MgCl2
- 50 uL total reaction
- 10 uL scaffold (111 nM)
- 10 uL working stock (520 nM)
- 5 uL 10x folding buffer with appropriate MgCl2 concentration
- 25 uL ddH20
- Placed in thermal cycler with Wei's 72HR2 program, labeled under "Adam (3-day)"
- 50 uL total reaction
Day 29 (2011-07-14)
- Worked on presentation for Yin lab meeting
- TEM-ed the nanoparticle chains
- Seem to be some chain-like structures, but could just be random
- Need to extract with gel
- Our homemade 5 nm gold nanoparticles look good though
Day 30 (2011-07-15)
Attended a one-day Microscopy Nanocourse, taught by Professor Jeff Lichtman.
Week 7
Day 31 (2011-07-18)
- Adam took out the Box 3 foldings out of the thermal cycler for me - the 21 mM MgCl2 tube, however, had melted
- Ran a 1.5% agarose gel of the 3rd folding of Boxwithlid
- According to Wei, one of the the two bands boxed in blue is my product - the bands are different from the M13 scaffold band
- Seems like there is no difference between 15 and 18 mM MgCl2, but Wei says that he has tried folding a 100-helix 3D structure in 15 mM before with no success - this box about 130 helices
- Excised the regions of the gel bounded by blue boxes, and purified them using Squeeze n' Freeze
- Nanodrop yielded Nucleic Acid A260 readings of around 0.1, so according to Wei, concentration should be okay
- Prepped 15 and 18 mM box foldings for TEM
- Under TEM of 15 mM, we saw a lot of unknown sperm-like structures:

- Under TEM of 18 mM, we saw a lot of seemingly cube structures, which Wei later dismissed as just bubbles:

- Will TEM tomorrow the purified samples with Wei
- Under TEM of 15 mM, we saw a lot of unknown sperm-like structures:


Gel Scan of 3rd Boxwithlid Folding
Day 32 (2011-07-19)
- AFM-ed the 18 mM Mg purified Box 3 samples from yesterday - in the bottom lane ("18B"), saw strange cubical dimer structures, but nothing conclusive
- Revised and added to the AFM Protocol
- Ran 1% agarose gel of the 18 mM Mg Box 3
- excised all of the non-staple, non-aggregate bands ("18 All")
- Squeeze n' Freezed the gel extracts
- Let sit remaining gel sit overnight in 0.5x TBE + 10 mM MgCl2 buffer, as per Wei's suggestion
- Made Box 4 foldings (al measurements in uL)
- 18 mM MgCl2: 25 water, 5 folding buffer, 10 scaffold, 10 working stock from B3
- 21 mM MgCl2: " "
- 18 mM MgCl2 + lock: 25 water, 5 folding buffer, 5 scaffold, 5 lock (100 nM), 10 working stock
- 18 mM MgCl2 + lock + key: 20 water, 5 folding buffer, 5 scaffold, 5 lock (100 nM), 5 key (1000 nM), 10 working stock
- Thermal cycler: Wei's 72HR program in 3A
- TEM the two 18 mM purified Box 3 samples - some structures, but nothing conclusive:

18T

18B - Amicon-concentrated the 18T and 18B Box 3 samples from yesterday
- Scanned Sherrie's gel

TEM of Bottom Band in 18 mM MgCl2 Folding of Box 3 - unknown structures
Day 33 (2011-07-20)
- Squeeze n' Freezed again the soaked gel (18 All) from last night, then Amicon-concentrated it
- Concentrated 18T, 18B, and 18 All are ready for TEM sample prep, but very little
- Worked on redesigning Wei's box for our purposes
- Changed both B and L caDNAno files so that they can be used with M13mp18 scaffold
- removed third layer from B, shortened L
- Two locking designs
- Azobenzene tweezer on outside of box: plus of easily access for key in strand displacement, negative of possibility of crosslinking between different Bs/Ls
- Today, identified lock staples for both B and L
- Azobenzene stretch-lock on inside of box: plus of B-L selectivity, negative of limited access for key in strand displacement
- Identify lock staples tomorrow
- Azobenzene tweezer on outside of box: plus of easily access for key in strand displacement, negative of possibility of crosslinking between different Bs/Ls
- Nanoparticle handles on both L and inside B
- Today, identified handles on L; tomorrow, do B
- Color scheme:
- Grey: normal staple
- Red: lock staple
- Orange: nanoparticle handle staple
- Purple: Needs 3' -TT
- Blue: Needs 5' TT-
- Need to create digital schematics
- Changed both B and L caDNAno files so that they can be used with M13mp18 scaffold
Days 34 and 35 (2011-07-21 and 22)
- Continued working on adapting Wei's box, finished the design, and ordered the strands... [comments below]
- Opening/Closing of the Box:
- Direct locking only - that is, every barrel has 24 staples on each end (12 times at each side) that 5' protrude with a sequence complementary to the sequence that 3' protrudes 12 times from the bottom-side of every lid (6 times at each end) - I realize this is confusing... will make diagram next week. But basically, I am scratching the azobenzene strand/sequence idea
- The lid lock strands will have a toehold, and I ordered in a separate tube the complement to the entire protruding part of the lid lock strands, in order to open the box
- Cargo Loading/Solubilization:
- No nanoparticle handles inside the barrel; just one/two nanoparticle handles in the center of the bottom-side of every lid.
- Two different sequence designs for the handle: one complements Nick's gold-conjugated DNA and has two possible protrusions, one complements Wei's gold-conjugated DNA and has one possible protrusion.
- Each protrusion has a toehold for solubilization; also ordered a set with PC spacers for alternative method of solubilization; also ordered the ordinary staples (no handles) to blank Wei's when Nick's are being used and vice versa
- Best Case Scenario: "two-pot, two-step" folding
- Opening/Closing of the Box:
- Fold lid and barrel in two separate "pots" - TEM/AFM these separately to show they exist
- Throw AuNP-DNA conjugates into the lid pot - TEM this pot to show that there is a nanoparticle associated with each lid
- Mix the two pots together - under TEM, show that the nanoparticle is now inside the box
- Blast the boxes with UV light or introduce the "unloader strand" - under TEM, show that the nanoparticle is STILL inside the box (now solubilized)
- Introduce the "key strand" - under TEM, show that the lids have separated from the boxes and that the nanoparticles have escaped from the boxes
Week 8
Day 36 (2011-07-25)
Photocleavage Experiment
- two strands
- "64cargo" = [20 bp - PC Spacer - 47 bp]: diluted today to 100 uM
- "PC Lock" = [15 bp - PC Spacer - 17 bp]: already at 100 uM, according to Sherrie
- created two thin-walled PCR tubes of each: 1 uL of the strand + 9 uL water
- cleaved one of each using UV lamp
Protocol Today Lamp Model Blak-Ray Lamp (Model XX-15, UVP, Upland, CA) UVP UVGL-58 Handheld UV lamp (Cambridge, UK) Wavelength 365 nm 365 nm Distance Between Sample and Lamp 5 cm 3 cm Shaking Yes No Time 5-15 mins 15 mins Power 3 mW/cm^2 ~53 mW/cm^2 = 6 W/(4*pi*(3 cm)^2)
- our power estimation can probably be further divided by 10 because of power lost through heat
- Here are some images of the reaction chamber used to suspend the UV lamp:






- ran a 10% TBE PAGE gel, 100 V, 1 hour, SYBR-Gold, 10 bp ladder
- photocleavage seems to work, but could have exposed to UV lamp for longer; and there seem to be many strands prematurely cleaved by ambient light
- extracted the uncleaved bands from the gel, and purified them using Squeeze 'n Freeze
- centrifuged twice through filter, second time with added water
- Repeated earlier experiment using these purified strands, with 5 uL of the purified strands + 5 uL water in each PCR tube, and exposing one of each to UV light for ONE HOUR, flipping over tubes midway through
- Here are the 10% TBE PAGE gels of this second experiment, 100 V, 1 hour, GEL BOX COVERED WITH ALUMINUM FOIL, SYBR-Gold, 10 bp ladder
- Success! The uncleaved lanes show very little premature cleavage, and the cleaved lanes show nearly 100% cleavage
Day 37 (2011-07-26)
- typed up adapted protocol for photocleavage
- played around with azobenzene strand, fluorimeter, and UV lamp with Adam and Yael
- raw data:
- results:

- helped Sherrie prep a couple of gels
- tomorrow:
- test if just adding one/two/three equator staples to folded open spheres (with equator staples missing) closes the sphere
- create pipetting map for box
- make diagram
Day 38 (2011-07-27)
- Created a Pipetting Map for our adaptation of Wei's box design: File:WeiBox Pipetting Map.xlsx
- Used Adobe Illustrator to draw a comprehensive schematic of our adaptation of Wei's box design
- Cannot post this schematic or the caDNAno files because the original design has not yet been published by Wei
- For closing the sphere, Dave Zhang suggested an idea: To first bind the strand displacement lock (F_n+) to each of its two equator staples, purify these three-strand complexes, and then add these complexes to folded open spheres (Han spheres that are missing their equator staples)
- This afternoon, we mixed these strands together in 100 uL reactions, using 1 uL of each of the strands (directly out of the plates, 100 uM) and Han's folding buffer
- We also had three controls: lock only, one staple only, and lock + one staple only
- These reactions were annealed for 75 minutes in the thermal cycler (Adam's NPRTF program)
- We then ran a 10% TBE PAGE gel on these reactions
- The gel staining quality is not very good
- We are not sure why the lock + one staple only lane looks identical to its analogous lane with both staples (there should be an additional band in the latter)
- Will run gel again tomorrow to rule out pipetting error
- Perhaps the two-staple complex runs similarly to three-staple complex
- The band we want should be the highest one
Day 39 (2011-07-28)
Sphere Stuff
- Sherrie ran a new gel of Dave's lock strands: staining turned out better, but bands are the same
- Excised the highest band in each of the three-strand complex lanes, mixed these bands together, and Squeeze n' Freezed: these complexes should now be at around 100 nM, assuming a 10x dilution because of gel purification
- Not sure if these bands are what we want because there is no difference between the 2-strand complex lane and the analogous 3-strand complex lane
- Sherrie looked up for me the equator staples in the sphere that are directly across from the hinge or 90 degrees from the hinge
- These are in the 6/23/11 Plate 2ab
- directly across = D5 and D8 = 12[50] and 11[420]
- 90 degrees away = C10 and D10, D3 and E2 = 14[201] and 9[111], 14[398] and 9[309]
- Formed three mixes of these:
- A = all of these staples (10 uL each)
- B = the ones directly across + one on each side = D5, D8, C10, E2 (10 uL each + 20 uL water)
- C = just the ones directly across = D5, D8 (10 uL each + 40 uL water)
- Each staple should be at about 100/6 = 16.7 uM
- Further 16x diluted an aliquot of each of these solutions to about 1 uM
- These are in the 6/23/11 Plate 2ab
- Testing Dave's idea, and A, B, C, as methods of closing the open sphere (missing equator staples)
- Assuming that ABC staples are at 1 uM, folded unpurified open spheres are at 10 nM, and Dave's locks are at 100 nM, and that we want a ten-fold excess of locks to spheres
- PCR Tube 1: 10 uL open spheres "O"
- 2: 10 uL open spheres with all equator staples added "X" (already verified that this closes the sphere)
- 3: 18 uL "O", 2 uL "A"
- 4: 18 uL "O", 2 uL "B"
- 5: 18 uL "O", 2 uL "C"
- 6: 10 uL "O", 10 uL Dave's locks
It's Box-ing Time!!!
- All the IDT oligos for Wei's box design arrived today, except for 47[103]_LL
- Resuspended all the tubes, except the photocleavable strands, the key, the and solubilizers, to 100 uM
- Using the Pipetting Map (File:WeiBox Pipetting Map.xlsx), created pre-stocks
- L:
- NP_N: 5 uL each of the 3 staples
- NP_W: 5 uL each of the 3 staples
- L_L: 5 uL each of the 12 (11 today, because missing one LL strand) staples
- L: 10 uL each of the 121 staples
- B
- B_L: 10 uL each of the 48 staples
- B: 10 uL each of the 142 staples
- L:
- Created working stocks
- L_N: 3 uL NP_N, 12 (11) uL L_L, 121 uL L --> 100/136 = 735 nM staples
- L_W: 3 uL NP_W, 12 (11) uL L_L, 121 uL L --> 100/136 = 735 nM staples
- B: 48 uL B_L, 142 uL B --> 100/190 = 526 nM staples
- Folded 6 tubes of origami: 50 uL reaction each
- 1: B: 10 uL scaffold, 10 uL B working stock, 5 uL 10x folding buffer (15 mM MgCl2), 25 uL water
- 2: B
- 3: B
- 4: L_W: 10 uL scaffold, 7 uL L_W working stock, 5 uL 10x folding buffer (15 mM MgCl2), 28 uL water
- 5: L_N: 10 uL scaffold, 7 uL L_N working stock, 5 uL 10x folding buffer (15 mM MgCl2), 28 uL water
- 6: L_N
- Placed in thermal cycler 4B, Wei's 72HR program, should be done Sunday at 4:30pm
- Brief meeting with Wei to discuss the box design
- He doesn't think we need so many lock staples on the B; thinks box will definitely close, but will it open?
- We also had a miscommunication with his gold handle; the L handle doesn't need a poly T spacer; the poly T next to the gold is meant to be unbound, with the rest of the sequence complemented all the way up to the L, like this:
binding - TTTTTT - Gold
binding - L
Day 40 (2011-07-29)
To Do:
See if ABCDave closed spheres (run gel)Conjugate gold nanoparticles to spheres (purify spheres first)
Purification of PC, O Spheres and SD, O Spheres
- 1% agarose gel, 2.5 hours, 80 V
- gel excision, Squeeze n' Freeze, and Amicon concentration
Testing of the A, B, C, and Dave's locks closing mechanisms
- 1% agarose gel, 2.5 hours
- None of these mechanisms seemed to have worked
Attaching gold to the inside of PC, O Spheres
- Amicon-ed one, non-gel purified, 50 uL tube of PC, O Spheres
- 100,000 MWCO, see paper notes for timing/speed details
- Amicon-concentrated the "3" tube of sucrose-extracted AuNP-DNA: 10k MWCO, first run through for 15 mins at 14,000 g, then two water washes for 10 mins at 14,000 g, then one inverted spin for 5 mins at 14,000 g
- Nanodropped the concentrated AuNP-DNA, UV-Vis 520
- 0.295
- 0.368
- 0.300
- 0.287
- average: 0.3125 = 116 nM, according to Steve's Excel worksheet
- estimated concentration of spheres to be at 5.68 nM = .5 yield after Amicon * 50 uL * 10 nM scaffold / 44 uL
- Mixed together at 2:16 pm in 20 uL reaction in PCR tube
- 2 uL 300 nM NaCl --> final concentration of 30 nM
- 6 uL sphere --> 1.5 nM
- 12 uL AuNP-DNA --> 69.6 nM
- approximately 45x excess of gold to spheres
Box Stuff
- 47[103]_LL arrived today, diluted to 100 uM
- note to self: never have Elizabeth order PAGE purified strands again because they are super expensive and because IDT gives you very little! (only 1.1 nMoles in this tube)
- added 3 uL of 47[103]_LL to L_L pre-stock from Day 39 (because used 22 uL of the 55 uL, 11-strand pre-stock yesterday)
- remade L_N and L_W working stocks, depleting the L_L pre-stock
- 3 uL NP-N/W, 12 uL L_L, 121 uL L
- Folded 4 tubes of origami as per yesterday's recipes
- 2 B, 2 L_W
- Used Han Buffer 2 (16 mM MgCl2)
- Put these 4 tubes in thermal cycler: !!!Shawn --> Wei72HR
- should be done Monday 7pm
Wei's Box Plan of ATTACK
Monday
1) Run ?% agarose gel of 100 uL of the B, 50 uL of L_Wei, and 50 uL of L_Nick
- Scan to check that the origami has folded (the scaffold band has disappeared)
- As gel runs, sit down with Wei and extract from him all protocols that are necessary for the below steps
- Excise the correct bands (ask Wei), Squeeze n' Freeze, and concentrate with Amicon filters (ask Wei for protocol)
- Nanodrop the resulting samples, using Nucleic Acid A260 (if between 0.1 and 0.4, concentration should be high enough for TEM/AFM)
2) AFM/TEM (4-8pm reserved) the B and the L separately to confirm their presence and their size
3) Following Wei's protocol, mix together aliquots of the B with aliquots of both L (all purified), let anneal overnight
4) Following Wei's protocol, mix together LN with Nick's AuNP-DNA, and LW with Wei's AuNP-DNA, let anneal overnight
Tuesday
5) TEM (3) and (4), determine yield
6) If (3) resulted in closed boxes, introduce key to see if the box can be opened (what excess?), let sit overnight?
7) If (4) resulted in L with attached gold, fold new L with photocleavable gold handles (3 days)
8) If (3) and (4) worked, mix an aliquot of (4) with an aliquot of B (as per Wei's protocol) to see if we can get gold into boxes
Wednesday
9) TEM (6), and (8) to determine successfulness and yield
10) Troubleshooting, etc.
Later
11) After L with photocleavable gold handles fold properly (7), see if loading of gold onto L and subsequent cleavage of gold off of L works
12) After (6), (8), and (11) are successful, test if solubilization works: load gold, close box, photocleave, check for leakage, open box
13) Celebrate!
Week 9
Day 41 (2011-08-01)
It's August!!! What?!
Some Things for Nick to do on Monday:
- Anneal AuNP-DNA to gel-purified AND Amicon-concentrated spheres (on Friday, we loaded the Amicon-filtered, no-gel spheres)
- Gel-purify and TEM the Amicon-filtered, no-gel spheres that have been loaded with gold
Ran a 1.5% agarose gel of first folding of Wei's box
- 33 uL of B in three lanes, 35 uL of LW in 1 lane, 35 uL of LN in 2 lanes
- Ran gel
- Squeeze n' Freezed, let soak in 0.5x TBE 10 mM MgCl2 buffer for 30 mins, centrifuged a third time
- Let remaining gel soak in more buffer overnight
- Amicon-concentrated (100,000 MWCO) the squeezed-out liquid: 14000 g 3.5 mins, then 1000 g 2.5 mins inverted
- According to Wei, no need to fill Amicon column to 500 uL when just concentrating
AFM unpurified boxes
- good images of LN but not of B
Nanodropped the Amicon-concentrated boxes with Nucleic Acid setting, DNA, A260
- B: 0.155 --> 1.55 nM
- LN: 0.236 --> 2.36 nM
- LW: 0.157 --> 1.57 nM
TEM
- saw purified Bs, purified L, unpurified L
- interesting that unpurified Ls lay on their backs, whereas purified Ls stand up on their sides
Took 2nd folding of Wei's box out of thermal cycler
Formed B-L complexes using purified samples, where Ls are approximately 2x to Bs
- B:LN
- 5 uL B --> 0.67 nM
- 6.5 uL LN --> 1.39 nM
- B:LW
- 4 uL B --> 0.5 nM
- 8 uL LW --> 1.05 nM
- Wei's "QDANNEA7" thermal cycler program
Folded more origami
- 2 tubes of B
- 10 uL M13 scaffold, 10 uL working stock, 5 uL 15 mM MgCl2 10x folding buffer, 25 uL water
- 2 tubes LW, 2 tubes LN
- 10 uL scaffold, 7 uL working stock, 5 uL folding buffer, 28 uL water
- the righthand-side PCR tube of each L is folded with the old USB "Control DNA"
- Wei's "72HR" thermal cycler program --> done Thursday evening
Days 42-47
- Experimental log from days 42-47 will be posted following publication of a manuscript by Wei Sun et al., from which we derived experimental protocols related to efficient AuNP attachment to nano-structures
Day 48 (2011-08-10)
- Shwinn used GE S300 to purify excess gold away from the LN:Au made yesterday
- Took out 8/6 origami folding, which includes LN_PC (PROTECT FROM SUNLIGHT)
- Worked on presentation for tomorrow's group meeting
Day 49 (2011-08-11)
Gave presentation at Yin Lab Group Meeting
Day 50 (2011-08-12)
Last official summer BioMod day! What an awesome summer! Adam took us out for ice cream!
School Year
2011-09-06
- made new Wei's DNA + 30x Au (commercial)
- folded two new 50 uL of LW_new
2011-09-15
- purified away excess gold from LW-PAu and LW-UPAu
- resuspended new handle staples for B to 100 uM
- unsuccessful TEM of LW:AuNPDNA
2011-09-17
- attempt again of LAu
- met with Peng
- B:LW-AuNP
- 20 uL 8/2, 8/4, 8/6 B
- 15 uL today's LW:AuNP-DNA
- QDANNEA9
- more LWnew:AuNP-DNA(3x) with Wei's salt, Wei's gold (pure), but with LW 8/6 mixed in (out of today's)
- QDANNEA9
- more LW_new
- 72HR
- next time: purify B, purify LW_new, TEM B:LW-AuDNA
2011-09-20
- gel-purified Bnew and Lnew
- 2% agarose gel, 1.5 hrs
- Squeeze 'n Freeze
- let soak in 200 uL 0.5x TBE 10mM MgCl2
- prepare TEM grid of 9/17 B:LW-AuNP for Wei to TEM later tonight
- made two 50 uL reactions of LWnew_PC, in thermal cycler 3A --> 72HR
2011-09-24
- finished Squeeze 'n Freeze of Bnew and LWnew
- Amicon 100kDa 3.5 min 14000g, 2.5min reverse 1000g
- Nanodrop: Bnew = 1.2 nM, Lnew = 3.7 nM
- mix gold with Bnew and LWnew
- QDANNEAN9
- took out LWnew_PC
- gel-purified (2 hrs, no light), let soak overnight after Freeze 'n Squeeze
2011-10-04
- Finished Freeze 'n Squeeze of LWnew_PC
- Amicon-ed LWnew_PC
- mixed LWnew_PC with gold, thermal cycler: GoldDec1
- Imaged BnewAu, LWnewAu
- purification of xs gold is good
- good yield of BAu
- could not focus LWAu well
- mixtures (3 uL + 3 uL)
- BAu +LAu
- BAu + L
- LAu + B
- ordered new strands: barrel handles with PC spacers
2011-10-08
- purified away excess gold from LWPCAu
- cleaved 10 uL of LWPCAu
- TEM is broken :(
2011-10-13/15
TEM
- LAuBAu - best yield of gold in boxes
- LPCAu - gold attached well
- LPCAu_cleaved (1 hr UV) - not many lids with gold
- mixed LPCAu with B: 10 uL B, 5 uL LPCAu
2011-10-20
TEM of B:LPCAu - couldn't find any gold at all

{kind=link}