User:Jszab:m13
Notes
g3 has an uncommon START codon: GUG.
According to the image in pDraw, there are a few groups of sequences:
Separate ones:
KanR
Tn903
Ori M13, portion 2
III, VI, V
Overlapping sequences:
M13 Ori portion 1 ,XI, I, IV
II, X
VIII, VII, IX
It is reasonable to expect that genes that share sequences perform similar tasks.
Indeed:
II, and X are both responsible for managment of dsDNA to ssDNA transition.
XI, I, IV - all are taking part in assembly
VIII, VII, IX - are all capside proteins.
I would not change the proteins responsible in assembly, given that they are membrane proteins thus having to obey more restrictive conditions. Also there is not much we can do except make it function, infunctional. We could use it for transporting other things than ssDNA of M13. However, there are plenty of other secretion system which serve other purposes that are more similar to our goals.
|
|
|
Overlapping: |
Genome engineering:
The most obvious proteins to engineer are those on the surface. We could do all sorts of phage display. So the first target for engineering are proteins: III, VI, VII, VIII, IX. We definitely should separate them. Ideally the whole genome would be to create a excisable promoter, terminator and ORF for each of the gene. So for each gene we would have to create 4 specific restrictions places.
However, since majority of our work will be display it might be enough to separate just coat proteins.
Single protein engineering:
| protein | function | reengineering idea summary |
|---|---|---|
| I | assembly | |
| II | replication of DNA + strand | Enclosing the gene II in maltose binding protein: http://www.jhu.edu/~cheme/ostermeier/pdf/19_JMB.pdf by random domain insertion. This would allow to control the rate of synthesis of + strand thus delaying replication until the right moment.This principle could be connected for example to quorum sensing [bacteria producing maltose?], thus creating a kind of bacterial "war" with suicide bombers: whenever a bacteria from another group enters in vicinity with its maltose - the cell releases phages. [I thought it would be a funny application.]. This approach would be probably faster than transcription induction. |
| III | phage tail protein (5 copies) | Deletion: we could create bacteria grain with extending filaments. Those, when covered with metal, or another material could create grains of a macroscale material [like portland cement which has similar molecular structure.] |
| IV | assembly | |
| V | binds ssDNA | This protein can bind + ssDNA. The gene should be isolated and we should check whether it can also protect from restriction nucleases, or does it increase stability of DNA. |
| VI | phage tail protein (5 copies) | |
| VII | phage head protein (5 copies) | |
| VIII | phage coat protein (2700 copies) |
- various phage displays: Proteins that will aggregate upon addition of a ligand. Upon mixing with a metal could create thicker nanowires than in prof. Belcher original publication. Mixing of various population interacting with each other could also produce waveguides which might be of great use for electronics. |
| IX | phage head protein (5 copies) | I would like to make a phage invading two pili at once. This could create a crosslinker between two cells. It would not be very rigid, but could be bolstered by covering with some materials. The problem is that phage wants to exit with p9 and p7. So a simple idea to create a single particle would be to create two populations of phages which has sticky heads. Upon mixing them we could check if the phages can join to two pili and bring two cells togather. However, it cannot be done with shaking. |
| X | DNA replication | Similarly as in pIII, but instead of deletion we could change the promoters for more or less efficient. This would allow to control the replication rate. If the pX is infunctional for a longer period - no + strands will accumulate and no replication will occur. Therefore, the smaller concentration should decrease the replication rate. |
| XI | assembly |
Systems engineering:
= Creating thick nanowires would make them more rigid. If we want to have definite shapes, we could align magnetic particles [like magnetosomes] in various shapes of magnetic field.
= See whether other type of nucleic acids can be packaged. Whehter modifications [methylation?] change packaging.
= See whether digestion of proteins inside the nanowire is possible - to create a thin metal shell.
= Evolve the p8 to bind light bending polymer [1], or another 'smart' biopolymer. When phages with deleted p3 are shedding from E.Coli host, external electromagnetic field would cause them to move. Since the bending polymer bends accordingly to polarization, the bacteria could be moving according to the lighting - thus we could control their movement.
Questions:
Make the protein neutral: No. The charge might be important for interactions among other protein.Moreover, the DNA which is directly
interacting with p8 is charged [negatively] which may confer the orientation of p8 regarding DNA molecule.
Encode all Leu with CTA: it might work, but probably will be significantly less efficiently transcribed. This is not the optimum codon for E.coli.
Double the size: In general No.Doubling the size of a protein would not probably be possible - given that the packing is dense. It was shown that without giving an abundance of WT protein no more then 10 residues can be incorporated. However, I could imagine incorporating the protein into a coat of a phage, for example by making p8 dimer - therefore it would be possible. In fact, this paper treats about ways of incorporating the big proteins into M13 phage coat, by making a fusion with p8:
S.S. Sidhu, G.A. Weiss and J.A. Wells, High copy display of large proteins on phage for functional selections, J. Mol. Biol. 296 (2000), pp. 487–495.
This paper states:
"Here, we show that the display of two different
proteins (22 kDa and 50 kDa) can be greatly
increased by simply mutating the P8 gene to which
the proteins are fused. This dramatic improvement
in display is likely due to better accommodation of
the fusion protein in the phage coat."
For p3
No. The protein has evolved negative for some reason. Maybe because of the charge on the cells which allows to invade them.
No. As above, for the same reason.
Double the size: yes, probably. the proteins are not densely packed, some additions were made so far in prof. Belcher's lab.
Transcriptional terminators:
2x stronger: yes
100x stronger: yes
2x weaker: possibly,with less efficiency.
100x weaker: no, it would lead to random lengths of transcribed proteins depending only on the processivity of RNA polymerase and luck.
Enterobacteria phage M13 sub tree [retrieved from: http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?mode=Tree&id=10870&lvl=3&lin=f&keep=1&srchmode=1&unlock]
-Enterobacteria phage f1 M
-Enterobacteria phage fd
-Enterobacteria phage ZJ/2
The tree one phylogenetic class above M13: http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?mode=Tree&id=10861&lvl=3&lin=f&keep=1&srchmode=1&unlock
In order to find the differences between phages I performed alignment with a few phages from vicinity in phylogenetic tree, and M13k07 sequence from 20.109 website:
http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nucleotide&val=9626232 - Enterobacteria phage Ike
http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nucleotide&val=2760297 - vibrio phage fs1
http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nucleotide&val=215394 - Enterobacteria phage fd strain 478
The rough analysis shows that the greatest homology exist between 1st and 6250th nucleotide among all the phages. There is no striking pattern within this span of sequence, however there are smaller and bigger consensus values. Occasionally clusters of slightly higher homology can be found. These are usually short and does not span over the whole protein - just a part of it. this suggests that some part of the proteins can be modified, however not necessarily whole proteins.
The IKE phage [third from the top] seems to have more DNA and creates gaps in other sequences. Sometimes so does fs phage [fourth from the top]:
 .
.
I could bet there is a greater homology from 700-800 which is in gene 10.
there is a high homology region around 2800-2900 which corresponds to gene 6.
More thorough analysis is necessary if we are really to find the highly conserved sequences. Also some work has been done already:
Peeters, B.P.H., Peters, R.M., Schoenmakers, J.G.G., Konings, R.N.H., 1985. Nucleotide sequence and genetic organization of the genome of the N-speciWc Wlamentous bacteriophage IKe. J. Mol. Biol. 181, 27–39.
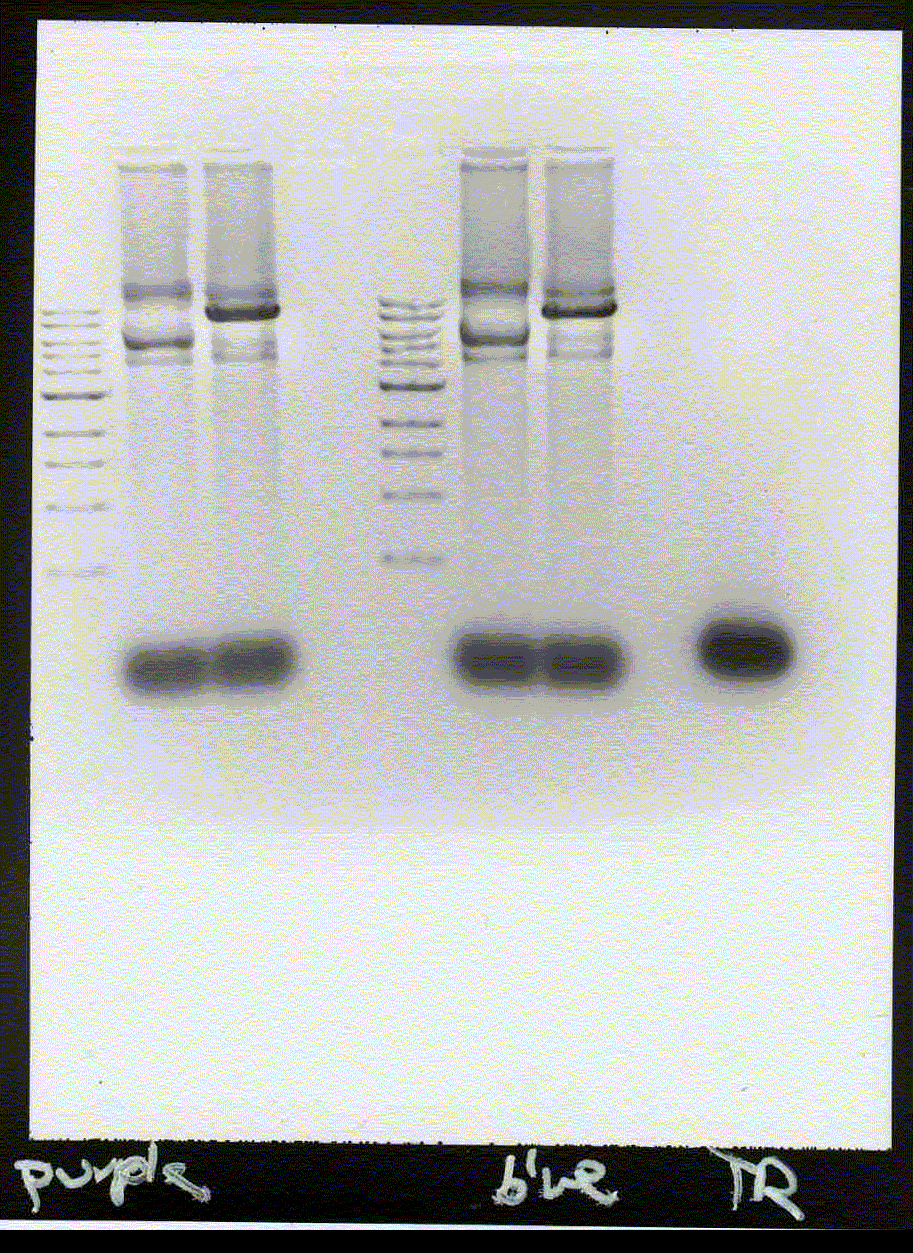
1) The band lies 10mm above 5kb, 16 mm above 3kb marker, 22mm above 2kb marker. This gives about 9.5kb if read from logarythmic plot.

2)0.01*10^12*10^-8 = 10 plaque forming units. Zero for DH5 strain. 3)
