Talk:20.109(S14):Introduction to cell strains and plating (Day1)
We will begin with a brief demo about sterile technique and how to use the tissue culture hoods.
Overview
Each team will be given two T25 culture flasks: one with CHO-K1 cells, and one with xrs6 cells. You will enzymatically detach these adherent cells, count them, and plate them at a specific seeding density in a 6-well culture plate. Next time, after these cells have doubled a couple of times, you will lyse them to make cell free extracts and begin your first assay of the module. For this reason, it is important that you take care to use sterile technique today.
It is essential that you do not mix up or cross-contaminate the K1 and xrs6 cell lines! We suggest that one partner is responsible for each flask, and that partners do not share Pasteur pipets or other equipment.
Protocol
- Waste disposal, preview: Pasteur pipets and pipet tips can go directly into the sharps mayo jars as you work. Please set aside serological pipets, conical tubes, tissue culture dishes, and gloves/papers; periodically put them in the benchtop biowaste containers or directly in the burn box.
- Each tissue culture hood is partly set up for you. Finish preparing your hood according to the demo, first bringing in any remaining equipment you will need, then picking up the pre-warmed reagents from the water bath, and finally obtaining your cells. Don't forget to spray everything down with 70% ethanol.
- One of the greatest sources for TC contamination is moving materials in and out of the hood since this disturbs the air flow that maintains the sterile environment inside the hood. Anticipate what you will need during your experiment to avoid moving your arms in and out of the hood while your cells are inside.
- Look at your cells as you remove them from the incubator. Look first at the color and clarity of the media. Fresh media is reddish-orange in color and if the media on your cells is yellow or cloudy, it could mean that the cells are overgrown, contaminated, or starved for CO2. Next look at the cells on the inverted microscope. Note their shape and arrangement in the dish and how densely the cells cover the surface.
- Aspirate the media from the cells using a sterile Pasteur pipet.
- Wash the cells by adding 3 ml PBS using a 5 mL pipet. Slightly tip the flask back and forth to rinse all the cells.
- To dislodge the cells from the dish, you will add trypsin, a proteolytic enzyme. Aspirate the PBS with a fresh Pasteur pipet. Then, using a 2 ml pipet, add 1 mL of trypsin to the flask. Be careful not to pull up the liquid too quickly or it will go all the way up your pipet into the pipet-aid!
- Tip the flask in each direction to distribute the liquid evenly. Incubate the cells at 37°C for 3-5 minutes, until the cells round up and are easily dislodged from the plate by tapping.
- While you are waiting, you may label the 6-well plate that you will share with your partner. Include your group color and today’s date on the left-hand side (assuming you are right-handed). Then label the two rightmost wells, which you will use for your cells, directly with the cell strain name and the initials of the partner who prepared those particular cells.
- Prepare two 15 mL conical tubes as well.
- Finally, this is a great time to clear out your trash!
- After retrieving your cells, add 3 ml of media to the trypsinized CHO cells and pipet the liquid up and down (“triturate”) to remove the cells from the plastic and suspend them in the liquid.
- Remember: Neither take up nor release quite all the liquid, in order to avoid bubbles.
- When you are satisfied that the cell suspension is homogeneous, transfer a known volume (such as 3.5 mL) to the appropriately labeled 15 mL conical tube. Take 90 μL from this solution into a (labeled!) eppendorf tube. Now keep the conical capped while you work at the microscopes.
- Add 10 μL of Trypan blue to the cells and pipet up and down.
- With a new pipet tip, transfer 10 μL of the dyed suspension to a hemocytometer, as shown to you by the teaching faculty. Keep K1 on top and xrs6 on the bottom, so you don't forget which is where.
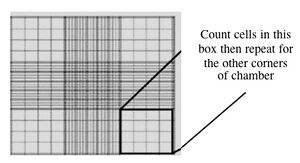
Counting cells using a hemocytometer - Count two diagonal corners, as shown to you by the teaching faculty. If the numbers are within 20% of each other, continue. If they are more different than that, count the other two corners. Be sure to record all of your raw data. Finally, take the average of either the two or the four values.
- This slide has an etched grid of nine large squares. The concentration of cells in a sample can be determined by counting the cells that fall within one such square and then multiplying by 10,000 to determine the number of cells/mL in the solution measured. (Always remember to convert from dilution to cell stock at the end!)
- Note that different squares are sub-divided into different grids. Very dense cells could be counted in the fine grids. In your case, the 4x4 grids and a 10x magnification will be most convenient for counting.
- Seed 200,000 cells in their dedicated well of the 6-well plate, and top off the media to 3 mL. Pipet up and down to mix.
- For example, if your concentration is 1 million (1 M) cells/mL, you would take 0.2 mL of cells and 2.8 mL of media.
- Finally, use the squeaky technique taught in pre-lab to distribute the cells evenly in the plate. Remember to move vertically and horizontally rather than in a circular fashion.
- Waste disposal, final: Aspirate any remaining cell suspensions to destroy them and then clean up the hood. Dispose any vessels that held cells in the burn box, and any sharps in the mayo jars or burn box according to the waste disposal preview above. The next group who uses your hood should find the surfaces wiped down, no equipment that you brought in left inside, and the sash closed. Please do leave the equipment that was already there.

