Sbb14-Christy Truong
Christy Truong 19:04, 13 February 2014 (EST)
Today we discussed which thermophile to purchase. We decided on thermotoga maritima and thermus thermophilus. Team 1 & 2 will handle thermus thermophilus and team 3 & 4 will handle thermotoga maritima.
Amino acids are split as follows:
Team 1 & 3: A C D E F G H I K L
Team 2 & 4: M N P Q R S T V W Y
Each teammate in every group is responsible for creating a construction file for 2 amino acids each. I have been assigned Methionine and Asparagine.
I was able to find Methionine-tRNA synthetase, but there were 4 EcoRI within the open reading frame. Thus, I plan to use a different restriction site, BsaI for digestion.
The enzyme that makes the asparagine tRNA is the same enzyme that makes the Glutamine tRNA, which is assigned to Tae.
20 February 2014
Here is my construction file for my methionine:
PCR oligo1/oligo2 on pBaD (1923bp, pcrpdt)
Digest pcrpdt (BsaI/BsaI, 12+16+1895, L, pcrdig)
Digest pBAD (NcoI/EcoRI, 3737+41, L, vectdig)
Ligate pcrdig and vectdig (Methionyl-pBAD) ----------------
Oligo1 Forward ccagtGGTCTCccatggccAAATTTTACATCACCACCCCAATC
Oligo2 Reverse ccagtggtctcgaattcTCAGGACACCTTCGCACCTG
25 February 2014
Today, as a group, we checked each others construction files and primers. The first line of my construction file was wrong and has been edited to:
PCR oligo1/oligo2 on thermatoga (1923bp, pcrpdt)
Digest pcrpdt (BsaI/BsaI, 12+16+1895, L, pcrdig)
Digest pBAD (NcoI/EcoRI, 3737+41, L, vectdig)
Ligate pcrdig and vectdig (Methionyl-pBAD) ----------------
Oligo1 Forward ccagtGGTCTCccatggccAAATTTTACATCACCACCCCAATC
Oligo2 Reverse ccagtggtctcgaattcTCAGGACACCTTCGCACCTG
27 February 2014
We finalized our primers during lab. Here is Group 4's primers: Media:BioE140LGroup4primers.xlsx
Today we made miniprep purification of our DNA. The goal was to purify plasmid DNA from cells (less than 100 kb)
The protocol can be found here: Arking:JCAProtocols
04 March 2014
Today we received our oligos and began making our PCR products.
For my first dilution I added 338 μL to my forward primer and I added 337.4 μL to my reverse primer.
06 March 2014
Today I realized I did my PCR product wrong so I remade another one, following the correct procedure this time. I did not put both oligos in one PCR tube. I had separated them, but now I understand why they need to be put into the same tube. That way, the section that I want to amplify will be cut out correctly. After running the gel, this image was obtained: 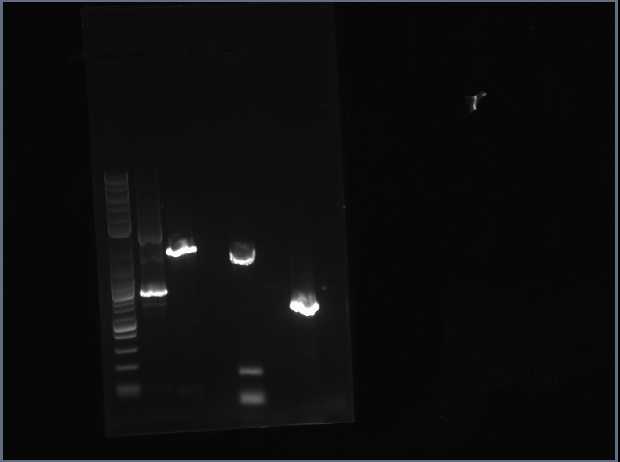
My PCR is the very last one on the right. As you can see, nothing is there so my PCR did not work. I had to redo my PCR again today. Hopefully it will work this time around.
11 March 2014
I ran my PCR according to the PCR protocol and put it in the thermocycler.
13 March 2014
Today I ran a gel on my new PCR and it worked. Please see picture below. In comparison to the ladder and other bands, my product is 1923bp and the band is at about 2000bp. I shared my gel with team 1. My band is the first band on the left. The other bands belong to Team 1. My band has high intensity and is a solid, fat band. My PCR worked.

This gel took the entire class period because we were having trouble with the voltage. Someone turned it off and we did not notice when we checked the progress of the gel. Next class, I will be prepared to run a digest on my PCR product and my vector.
18 March 2014
Today I was absent due to sickness.
20 March 2014
Today I performed my digest on my PCR product and on my vector. I prepped both solutions and set it in the thermocycler for 1 hour at 37 degrees Celsius.
While waiting for my digest, I made a gel using 0.3g agrose, 20 mL 1X TAE. I added 3 μL of gel green and poured the mixture into the mold and let it sit.
When it was time to load my samples, I added 2 μL of loading dye to my PCR digest and 6 μL to my vector digest.
My gel turned out well, but the computer generated a weird picture and it's hard to see my wells. However, this is the only picture I could produce. The first well is my Methionine, then my ladder, and the next three are my vectors.

01 April 2014
Today I did a ligation and transformed E. coli using the following procedure (different from the protocol listed in JC Anderson's website):
1. Thaw a 100 uL aliquot of cells on ice 2. IGNORE WATER ADDING STEP 3. Add 35μL of KCM to the cells 4. Put your ligation mixture on ice, let cool a minute or two (for Miniprep product, dilute by 10, then use 1μL of dilution) 5. Add 65μL of the cell cocktail to the ligation, stir to mix 6. Let sit on ice for 10 min 7. Heat shock for 90 seconds at 42°C (longer incubation may work better) 8. Put back on ice for 1 min 9. Add 100μL of 2YT, let shake in the 37°C incubator for 1 hour 10. Plate 70+μL on selective antibiotics, let incubate at 37°C overnight
I plated my methionine and a negative control on Ampicillin agar plates. I expect to grow many colonies on my plate, but much fewer on the negative control since those E. coli should not have functional plasmids with the Amp-R gene.
03 April 2014
I checked my agar plates today. Neither my methionine nor my negative control worked. I need to ligate, transform and plate again today. Here is a picture of my agar. As you can see, nothing grew.

08 April 2014
Today I ran an analytical gel on my PCR PDT to see if my product is still good. I ran 2μL of sample, 6μL ddH20 and 1μL dye. I got a really nice, solid bright band. To the left is my ladder and to the right is my PCR PDT. This shows that my PCR PDT is still good and full of DNA. I also redid a PCR in case the PCR PDT fails, but there is no need for that now.

10 April 2014
Today I digested my PCRPDT with BsaI and ran a gel. I did a PCR digest (PCR DIG 4-10) and a vector digest (VEC DIG 4-10). On the gel, my VEC DIG was 2nd to the last and my PCR DIG was the last column.

The gel on the right is not mine and is irrelevant. However, the gel on the left shows my bands. There were two bands for my VEC DIG and no band for my PCR DIG. That's not supposed to happen. I should have a strong band for my PCR DIG and a strong band for my VEC DIG.
Since they both fail, I decided to make a new PCR and start the entire experiment from the beginning, next class.
15 April 2014
Today I made an outline of what to do with Professor Anderson. I made a PCR, labeled PCR RXN.
17 April 2014
Today I ran 2μL of my PCR RXN on an analytical gel. My reaction is the 2nd one next to the ladder (first well). The rest of the wells belong to Tae.

I got a nice solid band so I went ahead and did a zymo cleanup on the remainder of my PCR RXN. Next class, I will do a digest on my pcr and my vector.
22 April 2014
Today I did a PCR digest with my PCR-DIG-RXN and my vector digest. I ran a gel on the right side with the vector being the last well and my PCR being the second to the last well.

Since I got good bands (my PCR has 2 bands because there is an internal restriction site and my vector is supposed to have two bands), I did a zymo clean up and I labeled my PCR: PCR-GP and my Vector: VEC-GP
24 April 2014
Today I did a ligation on on my PCR-GP and my VEC-GP and I made a negative control. I transformed by PCR and Neg control and plated them. Assuming I get colonies, I will do an inoculation on Tuesday, come in Wednesday to do a mini prep, and then on Thursday I can do restriction mapping digest.
29 April 2014
No colonies formed so I couldn't move forward with my plan. Luckily, Karim's project went very well and he had some leftover tryptophan and vector for me to redo a ligation and transformation, just so I can get some experience on a working experiment.
I had a little problem with the heat shocking step. The heat block wasn't at 42 degrees when I put my samples in. It was a mechanical error. As a result, my reaction may not work because the cells had a chance to warm up. I will find out next class.
01 May 2014
Success! I was able to grow colonies in both my negative control and my tryptophan. My negative control had two colonies and my tryptophan had more than 20+ colonies (please see figure below).

My next step is to incubate the tryptophan into four test tubes in a 37 degree shaker overnight.
02 May 2014
I got my test tubes back and I picked good colonies! I received cells on the bottom of my tubes.


This concludes the lab portion of my experiments. It was a great experience and I'm glad I took the class.
