Prof G. R. Desiraju's Lab
Drug Design and Discovery
Computational Approaches from X-ray Crystallography
Intermolecular interactions, especially hydrogen bonds, manifest themselves in somewhat similar ways both in crystals of small organic molecules and large biological macromolecules. We are trying to analyse crystal structures of biological macromolecules with respect to intermolecular interactions and to establish some general principles for their assembly using ideas and concepts of crystal engineering. The possible application of such a study is in the field of drug design. Where drug design is concerned, an overall knowledge is vital to obtain safer drugs. Especially, with the current importance of virtual screening, there is an urgent need to develop methods to efficiently study the chemical space of drug molecules. Our research aims at understanding the overall chemical nature of drug molecules through the study of databases of small molecules (drugs and non-drugs). In this respect there is a continuing effort in our group to develop methods for analysing drug-like and non-drug molecules.
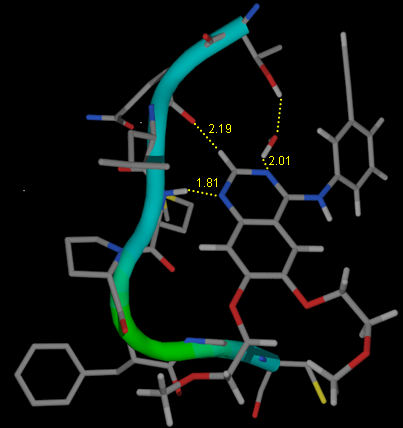
Traditionally drug design has relied more on serendipity than on rational design, especially in the early stages of lead molecule identification. But recent developments in the field of genomics and proteomics have forced the researcher to adopt more rational approaches to drug design to convert the huge amount of raw data into medicinally important information. We follow a dual strategy using both analog and structure based drug design based on the type of information available. In the analog based approach we use 3D-QSAR, pharmacophore generation and de novo design methods. On the other hand the structure based methods include molecular docking and molecular dynamics simulations to understand the basic nature of drug-receptor interactions. When the target structure is not known we try to model it through a computational approach (see illustration) Rational drug design with a flavour of strong and weak intermolecular interactions is being applied to design novel inhibitors targeting diseases like tuberculosis and cancer.
http://openwetware.org/images/c/cb/Drugdiscovery.png
Softwares : Hydrogen Bond Analysis Tool (HBAT) http://openwetware.org/images/6/61/HBAT.gif
Discription :- HBAT is a tool to automate the analysis of strong and weak hydrogen bonds present in a PDB file. This tool can be implemented in active site interaction analysis, structure based drug design and molecular dynamics simulations. HBAT is written using PERL and TK languages. It is a user-friendly desktop tool, which offers the user freedom to select several parameters. The program generates an MSOFFICE Excel compatible output file for statistical analysis, provides distance-angle distributions across various geometry ranges while tabulation of frequencies for each residue, ligand, water, and also nucleic acids can be done easily for any kind of interaction. Cooperativity in hydrogen bond geometry can also be studied in a defined region of structure.
link : http://202.41.85.161/~grd/HBAT.html
University of hyderabad http://www.uohyd.ernet.in/
last updated 18th August 2006
by Tejender thakur
email: tejenderthakur@gmail.com

{kind=link}
{kind=link}