Lab Notebook - Sherine
Back to Home Page
To All Notebooks
September
Wednesday, Sept 16
- Reviewing "Clotho Issues" list in prep for meeting
- Matlab and AI
- Meeting with Comp Team for Clotho; 5-8pm
Friday, Sept 11
- meeting
- running a Bradford protein assay to measure protein concentration
- currently looking at protocols for assay...
PROTOCOL
Thursday, Sept 10
- Prepared EspA cells for protein purification:
- added 10ml innoculated overnight culture into large flask, filled to 500mL with Amp LB
- placed on shaker/incubation for 3 hours (10:15-1:15)
- induced with arabinose (1:1000); induction time 4 hours, completed at 5:25pm
- Steps for protein purification
- pellet cells
- resuspend in lysis buffer
- sonicate, keep on ice; cool rotor to 4C
- 10mM, 3M or 300mM carbonate
- pour supernatant w/ resin and incubate 4C rocker
- spin to pack column, wash elute
- dialize overnight
Tuesday, Sept 8
- miniprepped first 2 colonies of each EB transfer
- restriction mapping: Order - C09ig0001-1, 1-2, 4-1, 4-2, 5-1, 5-2, ladder, 6-1, 6-2, 9-1, 9-2, Bca1092-1, Bca1092-1
![]() Clone 1 of all the E/B transfers were selected for use.
Clone 1 of all the E/B transfers were selected for use.
Friday, Sept 4
- Did a set of EcoRI/BamHI transfers for Jenn:
C09ig0001 into pBca9495AC vector, Lefty cell C09ig0004 into pBca9495AC vector, Lefty cell C09ig0005 into pBca9495AC vector, Lefty cell C09ig0006 into pBca9495AC vector, Lefty cell C09ig0009 into pBca9495AC vector, Lefty cell Bca1092 into pBca9495KC vecotr, Righty cell
- Pick colonies tomorrow (Sat), streak on AKC plates
- place in fridge on Sunday.
August
Thursday, Aug 27
- induced cells reinoculated yesterday
- CA-mgfp (what we know to be a good "positive" control)
- CA-1363 (neg)
- Spec-rfp (additional neg -- use for fluorescence comparison?)
- CASpec-mgfp-rfp
- CA-AIDA AtD no passenger (again, to see whether the AtD itself has "sticky" properties)
TOTAL volume: 7ml Cell culture - 1:10 dilution, 700ul per tube Arabinose - 1:1000, 7ul per tube LB with antibiotics - 6.293ml
- Placed in incubator at 10:15am
- With induced overnight culture - retry plate/comassie staining assay, except on a larger plate.
- Fluorescence assay - to be done in 96-well plates, where washes will not cause unwanted backgroud
Wednesay, Aug 26
We ran the plate/comassie staining assay and fluoresence assay for mgfp.
Monday, Aug 24
- Finished minipreps from C09ig0012 and C09ig0018 - 3 clones each
- restriction mapped (EcoRI/XhoI) all Comp team composite parts - correct!!!
- Parts C1-C3: Order c1-1, c1-2, c1-3, ladder, c2-1, c2-2, c2-4, c3-1, c3-3, c3-4

- Parts C4 and C5: Order c4-2, c4-3, c4-4, ladder, c5-1, c5-2, c5-3

Saturday, Aug 22
- Came in on Saturday morning to check on assembly plates Thien finished yesterday. Colonies were present. I picked 6 colonies of each, streaked them on ACK plates and placed them in 24 well block to grow.
- Checked for cotransformation of prior assemblies C1 - C09ig0033, C2 - C09ig0037, and C3 - C09ig0038. Miniprepped:
C1 - C09ig0033 clones 1,2,3 C2 - C09ig0037 clones 1,2,4 C3 - C09ig0038 clones 1,3,4
- C09ig0012 and C09ig0018 will be miniprepped on Monday. Restriction map for all 5 composites will be done then as well.
Friday, Aug 21
This morning, I first checked on plates for the assemblies, which all grew. Next, I checked the spec plates for M10046 and M10049 for cotransformation; miniprepped 2 un-cotransformed clones for each part:
M10046-2 M10046-3 M10049-3 M10049-4
- These, I restriction mapped as follows on the gel: M10046-2, M10046-3, ladder, M10049-3, M10049-4

- The parts mapped correctly. At 1pm, I started the Digestion portion of One-pot assembly with these parts:
pBca9495AC-Bca1363-1 with pBca9595CK-M10046-3 and pBca9495CK-M10049-3 to make Composite parts: C4- C09ig0012 (1363.M10046) C5- C09ig0018 (1363.M10049)
At 2:30pm, the Comp team came in for one final colony-picking session. This time, they picked 6 colonies from each of the three assemblies plates. The picked colonies were first streaked on ACK plates to check for cotransformation, and placed into 24-well block, with 3 ml KC LB.
After this, Thien stayed to finish assembling C09ig0012 adn C09ig0018. Ligase was added; the parts transformed into Righty cells for the final assembly; plated on AK plates.
Thursday, Aug 20
Before the rest of the team showed up, I picked colonies from re-transferred M10046 and M10049, which successfully grew this time! 4 colonies of each were picked, streaked on Spec plates, and placed into tubes with 3-5ml KC LB media.
At 10:30am, the rest of the team showed up - Nina joined us as well! We first checked the Spec plates to see if there was co-transformation in any of the 4 colonies that we picked from each plate. On average, about “1.5” out of 4 was cotransformed. We selected the cultures of 2 colonies that were not co-transformed for each plate, moving the culture into 2ml tubes to miniprep.
pBca9495CA-Bca1363, clones 1 and 2 pBca9495CA-Bca1092, clones 3 and 4 pBca9495AK-B09ig279, clones 2 and 4 pBca9495AK-B09ig283, clones 1 and 2 pBca9495AK-B09ig302, clones 1 and 2
Minipreps for these 10 samples were done by 12:30pm. After explaining the process of restriction mapping, we sat down to figure out which two enzymes to use to generate band sizes that can be easily resolved on a restriction mapping. For each of our 5 constructs, we concluded that digestion with EcoRI/XhoI would give us bands with sufficient size difference for us to tell apart on a gel. Expected band sizes:
pBca9495CA-Bca1363: 3308, 1196 pBca9495CA-Bca1092: 1985, 1196 pBca9495AK-B09ig279:2911, 1133 pBca9495AK-B09ig283:3622, 1133 pBca9495AK-B09ig302:3478, 1133
At 1pm I began setting up these 10 digestions for a 30min incubation period. At ~1:30pm, I placed these into the thermocycler, and at ~2pm, removed the tubes into the fridge.
2:10-4pm: MEETINGS (for both Comp and Wet teams)
~4:15: We met up again to run the gels!
We set up two gels, just so that the loading can be done more efficiently:
- GEL 1 Order (from left to right): Ladder, 1363-1, 1363-2, 1092-3, 1092-4

- GEL 2 Order: Ladder, ig279-2, ig279-4, ig283-1, ig283-2, ig302-1, ig302-2
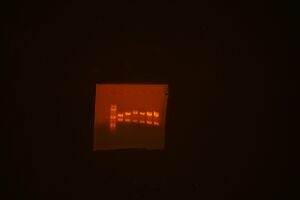
The clones that are correct and selected to use in assembly:
- 1363-1
- 1092-3
- ig279-4
- ig283-2
- ig302-1
Note: 1363-3 we save for a later composite assembly, once we get M10046 and 49 grown in media (starting today) and miniprepped and restriction mapped (tomorrow). We continue assembly as planned with 1092-3 and the three other parts.
At ~6pm, we set up the one pot digestion as listed in “Assembly Protocol.”
Lefty pBca9495AK-B09ig279 Lefty pBca9495AK-B09ig283 Lefty pBca9495AK-B09ig302 With Righty pBca9495KC-Bca1092
6:30-7:20ish biscotti/lunch/dinner with wetlab people… 7:30pm, added ligase to the three assemblies. Most of the team stayed with me for moral support until 8pm, when they left. At 8pm, Richard and I transformed the assemblies into righty cells, in preparation for the composite assembly, where they will be joined with lefty “pBad.passenger” parts. At 9:12pm, after a ~40min rescue, we plated the parts on AC plates.
Wednesday, Aug 19
Today was a relatively short day. This morning, I checked on our plates in the incubator. Every E/B transfer grew, except for M10046 and M10049; not completely sure why, but I started redoing the transfer for those 2 parts. This also means that we won't be able to assemble the composites requiring M10046 and 49, as they will be a day delayed.
The team came and picked colonies at a 11:30am - 4 colonies per plate, just so that we have extra clones in case a mishap occurs. We also checked for co-transformation on Spec plates. Using multiple colonies will also give us a bit of leeway, in case we have colonies that do end up being cotransformed. The colonies of the 5 continuing E/B transfers were placed into a shared 24-well block. Each well was filled with 3mL LB with correct antibiotics.
In Column 1 (downwards): Clones 1-4 of Bca1363 In Column 2: Clones 1-4 of Bca1092 In Column 3: Clones 1-4 of B09ig279 In Column 4: Clones 1-4 of B09ig283 In Column 5: Clones 1-4 of B09ig302
I also took some time afterwards to discuss "data analysis" schematics and the "One pot" assembly process.
Behold, my "If-Then Statements of Wetlab":
ECO/BAM TRANSFERS -if no colonies - REPEAT step -if yes colonies, proceed... COLONY PICKING, GROWTH IN MEDIA, AND CHECKING FOR CO-TRANSFORMATION -if no growth OR -if yes growth AND yes cotransformation - REPEAT step -if yes growth AND no cotransformation, proceed... MINIPREPPING AND RESTRICTION MAPS -if restriction maps failed - REPEAT step -if restriction maps correct, proceed... SEQUENCING (SOMETIMES OPTIONAL) -if sequencing incorrect - REPEAT previous step -if sequencing correct - Proceed to ASSEMBLY. END
Hopefully, tomorrow we can begin composite part assembly for some parts.
Tuesday, Aug 18
Starting today Joanna, Adam, Thien, and Richard will be coming in to do some wetlab work. The objective is for them to experience one round of composite part assembly, so that they can gain a better understanding of what computational tools would be helpful for similar wetlab processes. I will be working with them to coomplete the following 2-part, short term "project":
- Part 1 - EcoRI/BamHI transfers of basic parts into assembly vectors with double antibiotic resistances
- Bca1363 to pBca9495AC
- M10046 to pBca9495CK
- M10049 to pBca9495CK
- B09ig302 to pBca9495KA
- B09ig279 to pBca9495KA
- B09ig283 to pBca9495KA
- Bca1092 to pBca9495AC
- Part 2 - Assemble intermediate composite parts
- C09ig0033 {B09ig279.Bca1092} in pBca9495KC
- C09ig0037 {B09ig283.Bca1092} in pBca9495KC
- C09ig0038 {B09ig302.Bca1092} in pBca9495KC
- C09ig0012 {Bca1363.M10046} in pBca9495AK
- C09ig0018 {Bca1363.M10049} in pBca9495AK
The final composite parts (consisting of 4 basic parts) will be assembled on the robot, basically as permutations of C09ig0012 and 18 with C09ig0033,37, and 38.
The team came in bright and early this morning at 10:30am. Since they were orginally hoping to build basic parts (but we didn't have any that needed to be constructed at the moment...), I went through basic part construction conceptually. It was helpful to familiarize with the common terms found in basic and composite part making. Next, I described the Eco/Bam transfer process and some of the theory behind it.
Individual assignments:
Bca1363 and Bca1092 - Joanna M10046 and M10049 - Thien B09ig279 and B09ig283 - Richard B09ig302 - Adam
The schedule for the rest of the day is reconstructed below:
Starting Eco/Bam transfers: (protocol found on Eco/Bam transfers protocol page) -Set up digestion -Lunch (while digestion is running) -Ligation -Transformation -Plating Most of these were completed by 4:30pm!!
Intermittently, I showed the Comp team the various spreadsheets and organizational tools we use currently in wetlab, and discussed the importance of such organization - especially with high throughput projects that require the building of many permutations of parts. I should also mention that it was extremely fun learning sterile technique. Ah, the amazing uses of fire in the lab... We now wait in eager expectation to see if colonies grow on the plates!!
Monday, Aug 17
This morning, Susan and I compared our soluble gliadin assay data. We think that there may be a hit with espP(beta), because we saw the highest fluoresence readings from espP in both assays. However, data from Susan's assay on Saturday had in general lower fluorescence readings for all the samples. I ran another soluble gliadin assay with all the FitC-gliadin conjugate we had left in attempt to "confirm" that espP was a hit.
Important Notes:
- Since we only had ~300uL left, we planned to only try 30 samples, or duplicates of the 15 gliadin parts.
- accounting for mechanical error, the first set of 15 parts were placed with 100ul total volume per well of PBS+fitC. In the second set:
- Wells B1-9 have 100ul of solution.
- Well E2-5 have 90 ul of the solution.
- Well E6, containing espP(beta) displayer only received 60ul of solution.
- Results: all the fluorescence readings were very low, and the espP(beta) displayer that had shown high fluorescence seemed average to low in this assay. Also, since we only had incomplete "duplicates," we did not deem this assay completely reliable.
Aside from this, Susan is attempting another non-soluble gliadin assay. However, we are nearing the end of our gliadin assay experimentation, since there doesn't seem to be too much we could alter to make this assay give us more "definitive" answers. Our project focus is moving from foundational cell surface display to mgfp characterization, specifically.
Weekend, Aug 15-16
Didn't come in this weekend; Susan ran one more soluble gliadin assay and several more insoluble gliadin plate coating assays. Data to be discussed tomorrow.
Friday, Aug 14
Follow up on gliadin
"Quick and Dirty" assay:
*(Before: measure OD 600 of cells) *place 20ul? of gliadin in 100% EToH solution per well *allow to incubate at 37 so EtoH evaporates *place 5ul of cells + 95 ul of 1x PBS solution or 95ul LB(?) *incubate at 37 for 30 minutes *For consistency, use plate washer to dispense PBS washes and aspirate (program "GA100" runs this wash 3x) *Add 100uL LB to allow cell growth 3-5 hours; watch for cell cloudiness.
This particular run, the cells were incubated for 3 hours, with OD measurements every 30 min.
Note: This assay was inconclusive. The evaporated Gliadin-EtoH mixture caked at the bottom of the Maxisorp plate wells, so that OD measurements in the TECAN (which shoots like up from the bottom) were all the same.
Wet Q&D assay:
*measure cell OD? *place 10uL of gliadin/EtoH solution in wells *immediately (no evaporation needed), add 95ul of PBS + 5ul cell culture *incubate at 37 for 40 minutes *For consistency, use plate washer to dispense PBS washes and aspirate (program "GA100" runs this wash 3x) *Add 100uL LB to allow cell growth 3-5 hours; watch for cell cloudiness.
This was incubated for 4 hours, readings every 30 minutes. As of now, graphs are still being generated for this data...
Soluble gliadin-FitC assay:
Very similar to strep assay, except with FitC. *Take cell OD *pellet cells *resuspend cells in 1:10 dilution of FitC-gliadin and 1x PBS solution, so that each cell sample gets 10ul FitC-gliadin. *''(We are using 10ul FitC-Gli versus 1ul (like strep) because our protein concentration test via Watman paper+Comassie blue revealed that our FitC-Gliadin concentration was rather low. Most likely, we lost the protein in dialysis.'' *Aliquot 100ul of solution per well and resuspend *incubate for 30 minutes *continue as with strep assay, with 2-3 washes. *read final OD and fluorescence in TECAN - Optimal measurement perameters: **Measurement mode: Fluorescence **Select "Fluorescein settings" **use either "gain 5100" as with strep assay or "gain 19100"
The analyzed data can be found here.
Thursday, Aug 13
Two important things:
- dialysis prep for FitC-gliadin conjugate
- meeting on project
Wednesday, Aug 12
Today, I ran the strep assay for the mgfp-5 parts. Jenn and Gaby had prepared 2 different clones; both of which we ran assays on. Parts that did not make it into the assay:
Of both clones:
F09ig0266 {Pbad.rbs.prepro.StrepTag}{<mgfp-5>}{<CPG_L6>}(dblterm)
F09ig0273 {Pbad.rbs.prepro.StrepTag}{<mgfp-5>}{<TshA>}(dblterm)
General Observations and Notes:
- Uninduced samples of clone 1 were not used -- the cells were not placed in the well to be induced.
- Uninduced samples of clone 2 were used ONLY on the clone 2 plate. (The uninduced constructs should have been the same on both sets, just different colonies.
- No gunk was present throughout the entire assay
- At the 2nd wash, I noticed that the cell pellet was larger in the uninduced constructs.This may be because these two constructs were grown in glass tubes, and not in a 96-well block like all other constructs
- General fluorescence readings were low, with exception to two samples that had the highest average readings.
- Possible hits as concluded from data:
- mgfp-AIDA Atd - this had the highest average fluorescence, but interestingly, also had a lower OD (about 0.1, versus average 0.2)
- mgfp-upaG_short - also highest avg fluorescence, but 0.2 OD
- maybe mgfp-Hag AtD
All the possible hits had higher fluorescence readings on both clones.
The analyzed data can be found here.
Tuesday, Aug 11
We started working on the gliadin functional assay assay by first preparing a solution of 10mg/ml gliadin in 70% EToH and 10% acetic acid. The tube we placed on a rotating holder, allowing it to mix thoroughly overnight...
On a different note, I met with Adam from the Comp team today. I was asked to experiment with their new language called Eugene and with Spectacles, a program that allows you to visualize your "device" (ie composite part) as stringed "components" (ie basic parts) as you build them. Additionally, Adam and Lesia are in the process of writing a paper on Eugene. As such, we spent most of our time discussing various Design Scenarios that we hope to capture:
- Incremental Device Design - there can multiple constructs that use the same component or composite component; in wetlab this year, we have multiple constructs like this, of which I selected some for Adam to reference.
- Data Driven Design - Information gathered on properties of different components to help design more efficiently.
- Rules-based Design - I feel this is similar to Data Driven Design. For example, if promoter A and rbs B do not function together, this information will be relayed to the designer as a "rule". Supposedly, an error message will display relaying this information. The difficultly, I think, is that much of biology varies from case to case, and it might be difficult to pinpoint rules that "work" for a majority of the time.
Finally, I identified a list of composite parts with interesting features for Adam to use in Spectacles. We wanted a list of around 10 constructs that have the characteristics of:
- Homogenous - these constructs have the same re-occuring component or composite component used within the same device. They can be the same parts, or similar instances.
- Heterogenous - these constructs have varied components in the device; no/most components are not repeated.
In the list that I found, some of the constructs are representative of both homogen. and hetero - they can be used in either case.
Monday, Aug 10
Gliadin and scFv parts aren't s The KILR and INP - though they were parts we really didn't intend to make - looked absolutely amazing on the strep assay. The signals were the highest we'd ever seen. The readings were comparable to our intense positive control, and you could see the fluorescence by eye. KILR and INP are linkers/repeat regions that are around 30 aa long (?). After talking to Chris, we came up with the idea of attaching linkers to a bunch of our other parts that didn't have much signal. We could test to see whether these linkers help facilitate the moving of proteins to the surface for display. The plan is to include these additions with some of cellulases (hopefully), and make them with the internal passenger parts.
Sunday, Aug 9
Susan came in to run assays on the KILR and INP parts...data will be analyzed tomorrow.
Saturday, Aug 8
I came in at 11:20ish. Susan had induced two sets of final constructs: KILR and EILD. I induced the Cel5B and Cel9A sets, along with our positive controls and the negative control (in correct antibiotic media). Parts that did not make it into the final set out of the cellulase:
Cel5B -C1:pBca9495AC F09ig0165 -C3:pBca9495AC F09ig0171 -C7:pBca9495AC F09ig0169 Cel9A -D5:pBca9495AC F09ig0182 -D11:pBca9495AC F09ig0186 Refer to Susan's notebook for KILR and EILD constructs that did not get assayed.
Incubation period: 6:55pm-7:25pm
Important notes and observations:
- Uninduced constructs have 60uL cells in total 500uL volume
- Cel9A plate only has duplicates of the controls
- Before incubation observation: some wells had appearance of cell "gunk", yet again.
A good number of wells had this, but the wells that had most notable "gunk" were:
Cel5B: Rows ABC - column 1 Rows DEF - columns 4,5, and some in 6 Cel9A: Rows ABC - column 1 Rows DEF - columns 8-10, some in 3,4,6,7,12.
- After 2nd addition of PBS for wash 2, gunk was again observed:
Cel5B: Rows DEF - columns 4-6 Cel 9A: Rows DEF - column 6-10, 12; some also in 1-4; a bit was visible in rows ABC as well.
When we put the plates in the TECAN for fluorescence readings, most of the wells with gunk floating in them had high readings. We wondered whether the gunk was responsible for the high readings, so we experimented by taking all the gunk out of one construct's 3-well set. This was done to Cel5B plate - parts {Pbad.rbs.prepro.StrepTag}{<cel5B!}{<azo1653 AtD>}(dblterm) and {Pbad.rbs.prepro.StrepTag}{<cel5B!}{<CPG_L2>}(dblterm). When the gunk was removed, both times, we observed a dramatic decrease in the readings -- down to "background" level. Additionally, when the plate was shaken around and re-placed in the TECAN, readings of the "gunky" wells varied dramatically; for example the well containing {Pbad.rbs.prepro.StrepTag}{<cel5B!}{<CPG_L2>}(dblterm) varied from 100s to 400s. We think this is due to the position of the gunk at the time the fluorescence measurement was taken.
Complete data analysis for the all Cellulase parts can be found here
Friday, Aug 7
- Mgfp updates
Sequencing results came back this morning. All five samples came back with the mgfp part and Bca1363, looking perfect!! We we are extremely excited! As for my mgfp Eco/Bam transfers that were plated yesterday, I picked three colonies from each plate, and grew them up in AK media in a 24-well block. Hopefully, these will turn out just as well as the pBdr052 mgfp constructs did.
- Strep assays
Susan came in at around 8 am to induce the blocks. We are hoping to run 4 plates, maybe more, time permitting. Our first run will include Cel3A, Cel6A, Needle ScFv, and Gliadin scFv. Each of them are constructed with 13 different autotransporters. Some of the constructs didn't make it into the final set. These are:
Cellulases: Cel 3A: A9 - pBca9495AC-F09ig0147 Cel 3A: B3 - pBca9495AC-F09ig0154 Cel 6A: E9 - pBca9495AC-F09ig0198 Cel 6A: F3 - pBca9495AC F09ig0205 scFv: Needle A9: pBca9495AC - F09ig0009 Needle B3: pBca9495AC - F09ig0016 Gliadin C1: pBca9495AC F09ig0026 Gliadin C7:pBca9495AC F09ig0033
Basically, there are no constructs with autotransporters CPG_L2 and yuaQ.
Susan and I started the assays at around 3pm. Several important notes:
- ODs were kind of low, compared to previous runs - but, the positive controls were low too, we continued the experiment.
- After the 30min incubation, we pelleted cells and flicked the liquid out. However, in the process of doing so, we also lost some cell pellets. *Cell "gunk": resuspension was a bit difficult for some wells because they formed strange clumps. Interestingly, these were also some of the same constructs that lost pellets. We wonder if it might be a re-curring issue because of certain AtDs.Only the Needle scFv plate did not have this problem, nor the pellet loss problem.
- We "abandoned" Cel6A plate because of liquid contamination; we'll probably re-run Cel6A sometime.
From a brief glimpse of the data, we have some slightly higher fluorescence reads than we did with previous runs; but, they were still very slight differences. We will fully analyze the data when we've assayed the entire data set from all our final constructs. We will run more strep assays tomorrow.
Thursday, Aug 6
Mgfp updates
Susan and I came in this morning to miniprep our 8 new2 and new3 cell cultures (mgfp constructs). Unfortunately, 3-1 and 3-2 had a culture mixup, so we did not use those. In the end, we were left with 2-1, 2-2, 2-3, 2-4, 3-3, and 3-4; still a good number. These were mapped at around noon. The gel looked good for everything except 2-3. All 5 colonies that looked good were sent for sequencing. Even though the gels look good, we're still not 100% sure that it means 1363-mgfp is completely there; last time (refer to Jenn's notebook), the gels for this construct also looked "good", but since the mgfp part is 200bp, it is very difficult to resolved whether the mgfp is present compared to the 1363 part. Only sequencing will confirm. While we're waiting, I am doing Eco/Bam transfers for all 5.
Picture of the gel: At the left of the ladder are the constructs cut with BglII/XhoI; on the right are EcoRI/BamHI cut constructs. The constructs themselves are in order: 2-1, 2-2, 2-3, 2-4, 3-3, 3-4.
Internal Passengers
These looked a little strange on the gel, some with more bands than there should be. We are currently running Eco/Bam analytical digests on the basic parts themselves, just to make sure the parts are there. It could be that the composite parts are the problem.
Assays
We talked about the assays that we'll be doing these upcoming days. Susan and I will be responsible for the strep assays and the gliadin assay (when the gliadin components come in). Tonight, Jenn will be spotting colonies into fresh media, and Susan will induce early tomorrow morning. Tomorrow will be Assay Madness - Part 2!
Wednesday, Aug 5
I came in at around 9am to check our mgfp plates. Both had very few colonies, which were also kind of small. We waited until later today to pick colonies. In total, I picked 4 colonies from "new 2" and 4 colonies of "new 3".
Additionally, Susan and I learned how to do 8 strip minipreps - for the Eco/Bam internal passenger constructs. It was quite the experience - particularly when we were trying to remove all the supernatant from the cell junk without accidently pipetting out the cell junk too! Now, we are enlightened about this wonderful art... We will run a gel for these tomorrow morning.
Tuesday, Aug 4
Mgfp portion:
Susan came into lab at around 4:30a.m. (!!) to pick colonies and streak on Spec plates to check for co-transformation. She picked 3 colonies of each (of the "old" and "new" mgfp constructs). Colonies 2 and 3 of the "old" and "new" mgfp constructs were miniprepped today at around 5pm. Restriction mapping results are yet to come. Given that those look "fine," we'll go ahead and assemble them by hand while waiting for sequencing results.
At around 6:30pm, we determined that the colonies of the "old" construct looked strange, so we decided not to use these. However, colonies 2 and 3 of the "new" construct looked fine - these we used both and assembled with pBdr052-pBca1363. The protocol used is as follows:
Digestion: 1uL lefty plasmid(methylated) 1uL righty plasmid (methylated) 1uL NEB2+ATP 0.3 uL XhoI 0.3 uL BglII 0.3 uL BamHI 6.1uL Water Digest for 1 hour at 37C + 20 minute heat kill at 65C. Add 0.3 ligase to each sample. Let sit on bench top for 30 minutes. Transformation is done as usual. Rescue was done for 40 minutes, and 70 uL were plated.
Susan stayed until 10pm to plate.
Eco/Bam transfers of internal passenger contructs
The colonies looked really small this morning, so we'll have to wait until later in the afternoon/evening to pick colonies. Jenn ended up doing these at 8:45p.m. They will be miniprepped tomorrow.
Tonight was also our wetlab movie night!
Monday, Aug 3
Gaby will be miniprepping 16 the E/B transfers today. They were restriction mapped in the afternoon - at around 4pm, we determined that they looked great! Assembly for the lefty and righty E/B-ed can now move forward.
Susan and I have been assigned an additional "new project"! Jenn found out that the mgfp parts didn't assemble properly with Bca1363 (pbad...strept-tag Lefty part) - sequencing results showed that there was only Lefty vector. We're going to attempt re-assembly by hand. We currently have the mgfp basic part (B09ig239) gatewayed into assembly vector pdr052.
Procedures:
- transform from plasmid into pir Righty cells (include the plasmid from previous trials that failed)
- pick colonies; streak on Spec plates to check for co-transformation
- miniprep, d-map and send for sequencing
- Assemble by hand with Bca1363
Today we started the transformation portion of this.
We are also doing E/B transfers for 21 internal passengers constructs. These, we started digestion at around 4:15pm. The protocol for these is as listed in the E/B transfers page.
Sunday, Aug 2
Didn't come in to lab today, although Susan and I were thinking of started the 2nd round of E/B transfers for the inner constructs. We'll do those tomorrow morning instead. I also heard that the plates from yesterday didn't look completely wonderful, with possibility of contamination. Colonies were picked from these plates anyhow, and will be miniprepped and D-mapped on Monday.
Saturday, Aug 1
In the afternoon, Susan and I transformed and plated 16 <displayer><dblterm> constructs that were Eco/Bam transferred earlier in the day. Colonies will be picked tomorrow. We may start a second round of E/B transfers.
July
Friday, July 31
Jenn is finishing up Eco/Bam Transfers for the intermediate constructs that did not transfer well the first time/left over. The positive control 9494 is also being moved from vector pBca9145 into pBca9495 for consistency purposes in our strep assays.
We went through a To-do list for tomorrow:
1. miniprep EC100D transferred parts (+ analytical digest)
2. E/B transfer the minipreps into regular righty cells.
Thursday, July 30
Nothing much going on today. iGEM BBQ at Tilden Park.
Wednesday, July 29
The data we got from last night's assays were very unpromising. Aisde from the positive controls that had very high signal, everything else seemed to have little to no flurescence signal; nothing was higher than "background," with signal as low as the negative control.
The data we received from OD measurements can be found here
Data from the fluorescence measurements can be found here
We came up with several reasons why this happened:
- the assembly vector pdr052 contains the nonfunctional phagemid device that is also under a pBad(arabinose induced) promoter. The RepL region is most likely toxic to the cell; that would explain why expression level could be low.
- the strep assay is not able to "catch" everything expressed on cell surface
- our positive controls were really bright most likely because they are in high-copy plasmids; all the composite parts are in lower-copy plasmid under pir gene regulation.
Because of the toxic part in the assembly vector, we plan to Eco/Bam transfer the <pbad...streptag><passenger> constructs and <displayer><dblterm> constructs to the pBca9495 assembly vector. These will be high copy plasmids, without the pir gene regulation and toxic phagemid. Hopefully, we can get better signal from the final parts if we reconstruct these intermediate constructs. This will set us a week backward, but we hope that this will allow us to get more reliable data from the strep tag.
Tuesday, July 28
My two 9494 and 1363 in EC100D plates grew. They are now going to be kept in the fridge, in case we want to grow up liquid culture from the colonies. I am pondering the need to miniprep and restriction map these??? It probably is not necessary...
Susan came in early this morning to induce all the cell cultures and controls we plan to assay today. We should have 4 plates to assay including triplicates. Terry also brought to our attention this morning that we should keep the "old" pBca9495-Bca9494 positive control, because we know for a fact that it fluoresces. Given an experiment where nothing glows (and goodness gracious we will never see that happen...), as long as we have a control that "for sure" works, we can more easily identify what might have been the problem. If that positive control doesn't glow, we could conclude that something was wrong with the streptavidin binding.
As of now, here are the controls we plan to use for each strep assay:
- Positive Controls:
- pBca9145-Bca9494 in DH10B cell-line{AraC-Pbad}{rbs.cpx} (this also contains a strep tag and has been tested for display)
- pBca9145-Bca9494 in EC100D cell-line
- Negative controls:
- pBca9495CA-Bca1363 in EC100D
- pBca9495CA-Bca1363 in EC100D
I have also updated the protocols with the most recent information that we have collected. We are still hoping to get better data on the Growth Assay as we run the assay with our composite parts; some changes may be made and some procedures (ie. whether to induce for 5 hours before TECANing or inducing overnight...) will be confirmed soon.
Strep and Growth Assays - 3:30-9:00pm
- Strep Assay - 4 sets
- Cel5B
- Cel9A
- Needle scFv
- Gliadin scFv
- 3:30pm - Susan, Patrick, and I planned out what should be on each of the 4 plates
- 5 controls in triplicate = 15 total
- +pBca9145-Bca9494 in DH10B
- +pBca9145-Bca9494 in EC100D
- +pBca9495CA-Bca1363 in EC100D
- -pBca9495CA-Bca1363 in EC100D
- -pBca9145-Bca9494 in EC100D
- 16 constructs in triplicate = 48 INDUCED
- 3 constructs in triplicate = 9 UNINDUCED
- All extra wells filled with PBS
- 5 controls in triplicate = 15 total
- 4:30-8:30 - Susan and I ran the Strep assay protocol listed here....
- 8:45-9:00 - I set up the Growth Assay for the Gliadin scFv plate
- All the outer-rim wells are filled with PBS; EXCEPT for A2-A7, which have triplicates of induced and uninduced contruct F09ig0018.
- 16 gliadin constructs x3 = 48 INDUCED
- 2 controls, each in triplicate; in rows B-D, columns 6 and 7 respectively
- pBca9495CA-Bca1363 in EC100D (uninduced)
- EC100D without plasmid (uninduced)
- the same uninduced set (F09ig0018)in wells A5-A7 was duplicated in rows B-D column 9.
Susan put this plate in the Tecan at around 10p.m.
Monday, July 27
A set of Strep and Growth assays were done over the weekend...but both looked a little strange. Added to the fact that we did not have controls, the data was not very informative. Otherwise, from the image, it seemed like there was very little, if any fluorescence. Today, I basically set up for a re-run of the assays, scheduled for tomorrow. Particularly for the Strep Assay, I prepared positive and negative controls 9494 and 1363 by transforming them into EC100D cell-line, the same cell-line that all our composite parts are in (it has the "pir" gene imbedded in the genome). We also have a negative control for the Growth Assay, which will be just the EC100D cells without a plasmid. Having the controls in the same cell line is for the matter of consistency.
Cocktail:
- 30uL KCM
- 50uL Water
- 200uL EC100D cells
Transformation and Rescue as listed in Protocols.
9494 and 1363 samples were split between liquid media and plating. For both, a total of 100uL was used: 50uL was placed directly into LB with corresponding antibiotics, the other 50uL was plated on corresponding antibiotic plates. From the EC100D sample, 100uL was placed into LB - nothing was plated. Earlier, Jenn mentioned that there might be something fishy with the current EC100D strain and that a new batch was being made; as such, it wouldn't be necessary to keep a plate of the old strain. Meanwhile, since we want to continue with our assays, we will use the current EC100D strain.
At any rate, these controls should be ready to use tomorrow when we run both assays.
Since we had a set of re-inoculated cells - the Cellulase parts and SOD+scFV parts - from Sunday, we decide to measure the ODs. Apparently, on Sunday, the induction/innoculation processes were not long enough? so there is still some experimenting going on right now with this.
At the end of the day, the cellulase parts and SOD+scFV parts were re-reinnoculated for tomorrow. We also decided that we should have a positive control in a high-copy plasmid with the pir gene. At this point, 9494 is being EcoRI/BamHI transferred into said plasmid. We will most likely use this control if it seems like we are getting no fluoresence from the assays - this is assuming that the lack of fluoresence is because of low expression.
For the rest of this week, we are hoping to do Strep assays during the day (we could probably go through a good number, since each plate requires minimal time in the TECAN)and Growth assays in the evening, as that takes 6hours at a time. Everything should be assayed in triplicates, so we'll be going through lots of plates...
In other news, this is most likely the longest entry I've written this summer! More to follow as we journey into the phase of Assay madness...
Wednesday - Friday , July 22 - 24
Friday
- continuing a bit of research on caspases - recombinant caspases, and other caspases' inhibitors
- learning/finalizing the Growth Assay protocol from Tom
Thursday
- Finished hunting caspase 3 inhibitors - there are at least 5 that I found that seem kind of promising...
- Meeting
Wednesday
- Looked at "miniature" assays for parts of the signal transduction scheme
- Movie night at UA7
Tuesday, July 21
I identified a list of Caspase-3 inhibitors that I want to look up. This will be my "project" for most of the week this week. Should be interesting!
Monday, July 20
The gels from Sunday still looked a little strange - particularly everything in Lefty cells. So, 4 of us are running experiments, hoping to identify what is the issue:
- is it the enzyme (BamHI) that's not cutting properly?
- is it the minipreps from the robot that aren't clean enough?
- is there something wrong with the lefty strain itself? (transforming "good" parts into lefty to check)
- are the composite parts there?
We came to the conclusion that everything else was pretty much fine except for the minipreps. The parts are all there from the minipreps done by hand. Most likely, the minipreps from the robot have not come out as clean as we would like them. Chris thinks that possibly the detergent (or other substance) not washed completely from minipreps might have interfered with BamHI restriction activity. It seems like that is the case...
Sunday, July 19
- Helped insert expected band sizes for BamHI/XhoI Dmaps.
- Transform confirmed "good parts" from previous sets into pir-Lefty strain. This is to check whether our current parts are causing the pir-Lefty cell issue, or whether there is something inherently wrong with the pir-Lefty cell strain.
Week of July 13-17
Research for Signal Transduction continues... A couple other highlights:
- July 16 - official photoshoot...
- July 17
- added two new deubiquitinating enzymes to our list of parts
- Jen discovered a number of issues with assembly -- the gels were not turning out correctly. The weekend will be spent trying to trouble-shoot these issues...
Friday, July 10
I've hopefully found one "mutant TEV -type" protease...
Thursday, July 9
We had our weekly Wetlab meeting: we are just starting a second round of research for basic parts. I will be researching for the signal transduction project. John caught us up on all the prior research he's done on this.
Wednesday, July 8
- meeting with Doug and Thien
- movie night #1 with Comp team
Tuesday, July 7
- Helping out with Strep Assay - Photographing portion: we measured OD for M10210-M10225 in the Tecan, with samples placed in a V-bottom plate and a flat bottom plate. When initializing "shake" function, we found that "orbital" pellets the cells to the bottom of the V-bottom; instead, we decided to use "linear" shaking, to more effectively mix the cell culture.
- set up overnight cultures for 9494 (pos control) and 1363 (neg control)
- running assay for parts A1-A4 (which are strep tag - displayer constructs), along with pos and neg control
- Clotho plate manager mystery solved, with the help of Doug.
- preparing notes for meeting with Doug and Thien to discuss Clotho issues
Monday, July 6
Nothing much happening today besides Social event day -- Bowling! I am also now helping with the Strep assay - photography portion.
Sunday, July 5
Sequencing results for ig 280, 282, and 283 have come in. Tube switch between 280 and 282 was confirmed; but both sequences are "Perfect". 283 maintained same missense mutations, which means they probably existed in the original template -- deemed "Perfect".
Friday, July 3
I'm making a list of things that should be modified on Clotho. It is in the making. I also came across a TUBE SWITCH (!!!!) between ig 280 and 282.
Thursday, July 2
I analyzed two of Elicia's parts (Eig55 and 56, which are ig277 and ig324, respectively). ig 277 was perfect, and ig324 was a perfect partial. All minipreps that have come back successfully have been entered as "DNA" parts in the Clotho Parts Manager. Boxes are still being re-organized, particularly the "PCR products" Box, which desperately needs to be updated constantly.
At our meeting to discuss progress, we caught up on what everyone was working on. We're also planning for me to meet with Doug (comp team) and see how Clotho can be improved to include information that I'm otherwise placing on a google-spreadsheet. It would nice, if, by the end of the summer, we could wean ourselves off of google spreadsheets.
Wednesday, July 1
More organization ensues... We have added a "Waiting for seq results" box, as well as a box for all PCR products that are either fails or are not needed(ie we have minipreps from them already).
June
Tuesday, June 30
All the tubes are now organized and placed in 6 different boxes, according to category and color.
- PCA/PCR Primers - RED
- PCA assemblies - BLUE
- PCA amplifications - GREEN
- Purified PCR products (gp and zymo) - YELLOW
- Miniprepped samples - WHITE
- Digested vectors/templates - cardboard box
There is also a box for some things that we don't want to throw away immediately because they "may" be useful later. Those are kept in the "FULL" box.
Also currently working on: Clotho data entry: part-vector
Monday, June 29
I started re-organizing our freezer boxes, and putting information on a corresponding spreadsheet. Our goal is to have all the tubes in the boxes color-coded, and easily match them on a giant spreadsheet. The giant spreadsheet will have all the parts that we have built so far (basic) and have most, if not all relevant information regarding each particular part.
Wednesday, June 24
Ran an analytical gel on ig213,239,249, and 261 this morning. The bands for 249 and 261 look fine, although 213 and 239 look a little faint. For 239, it might have been just because the part is small. For 213, the concentration might have been a bit low. Patrick is re-eluting ig 213 and 239 with 8uL (instead of 50uL) water. Hopefully, the increased concentration will work better.
Aside from transforming our 4 synthesis parts, Patrick and I also re-ligatead and transformed all 12 synthesis parts from the day before. The colonies from those 12 didn't look too promising, so re-ligated them. If that does not work, we will mostly re-digest them from the beginning.
Tuesday, June 23
Oligos for Project B09ig277 came in. I'm working on the 4 synthesis parts with Patrick:
- ig213
- ig239
- ig249
- ig261
We assembled, amplified, and purified them. Will run an analytical gel on it tomorrow.
I also helped Tom, Elicia, and Joey with matching part numbers, oligos, ect...for their 10 basic PCRs. To make life less confusing in the future, I've also started a spreadsheet that contains all the necessary information for PCRs. I am hoping to do one that organizes oligos and basic parts as well.
Monday, June 22
Morning - Analytical Gel: everything looked correctly sized
Digested parts:
ig1 ig27 ig57 ig79 ig113 ig121 ig129 ig137 ig145 ig171 ig201 ig207
Analytical gel for digests: Gel 1: looked good Gel 2: smaller parts looked faint - re-digested ig129, ig137, ig201, ig207.
Gel zymo:
- ig1,27,57,145, 171 were zymo-ed and eluted with 8uL water.
- Three vector samples were eluted with 10uL water.
- ig79 was eluted accidentally with 16uL water.
- ig113 and ig121 are <300bp, but did not have isopropanol added before the first spinning. We took the liquid that was spun out, replaced it in the column, added 250uL isoporopanol to each sample, and respun them. Then we continued as usual with Zymo protocol.
Ligation: Everything ligated as usual, except for ig79. 2uL of insert was used instead of one, because of 16ul elution in zymo step.
Transformation: Same as usual, except we used 70uL of cell cocktail instead of 75uL.
Plating: Instead of using all the cells for each sample, we took only 75uL.
Friday, June 19
-oligos arrived
-set up PCRs for 12 synthesis parts
Finished adding parts for corresponding ordered oligos:
| Number | Part |
| B09ig302 | {<VirG AtD>} |
{<VirG AtD>}
{<yuaQ AtD>}
{<AIDA-1 AtD>}
{<SOD>}
{<Tir-M>}
{<cel3A!}
{<cel5B!}
{<cel9A!}
{<cel6A!}
{<upaG_short!}
{<CPG_L2!}
{<CPG_L6!}
{<espP(beta)>}
{<ehaB>}
{<eCPX>}
{<TshA>}
-Parts double-checked.
I believe that completes our first set of goal parts.
Replace part nickname in Database
Thursday, June 18
Several of us fiddled with the TECAN plate reader and planned the protocols for the streptavidin tag assay, as listed here. I inserted several more parts (corresponding with ordered oligos) into the Clotho Plate Manager:
{<caspase 3>}
{<mgfp-5>}
{<Tev N>}
{<Tev C>}
Wednesday, June 17
Second batch of oligos got ordered. We decided that the cyanide pump probably doesn't exist, so cyanide degradation/bioremediation is officially off the list. I also entered our basic parts for the corresponding oligos that were ordered (6/16 and 6/17) into Clotho :
{<azo1653 AtD>}
{<Nmul_A1566 AtD>}
{<OprF AtD>}
{<cl02365 AtD>}
{<VtaA11 AtD>}
{<Hag AtD>}
{<Pcryo_1225 AtD>}
{<Hia AtD>}
{<type IIIs Needle Complex scFV>}
{<gliadin binding scFv>}
{<Cub>}
{<Nub>}
Tuesday, June 16
We're finishing the "research phase" of things. The first set of oligos got ordered. We also ran an streptavidin tag assay today. In other news, I'm hunting for a cyanide efflux pump...
Monday, June 1
iGEM 2009 begins -- this should be an awesome 2nd year (for me)!! With the invivo Gateway that was started by iGEM 2008, we are planning to use the robot to help us make numerous constructs. A part of last year's project still carries through to this year - which is quite fantastic! At the moment, lots of project ideas are being tossed around, discussed, and researched.
