Haynes:FCMamalian FluorGene
Detecting Fluorescent Protein Expression
Karmella Haynes 2014
REAGENTS:
- FACS Buffer - 1% FBS in 1x PBS (stored at 4°C)
MATERIALS
- FACS tubes with strainer caps - 6 mL capacity (EMS 64750-25)
PROTOCOL
This procedure is for two samples: one fluorescence-expressing experimental sample and one non-expressing "blank" control. Scale-up reagents as needed for more samples.
Harvest the cells - do the following in the TC room
- Obtain an ice bucket. Aliquot 10 mL FACS buffer (5 mL per sample) into a conical tube. Chill on ice.
- Harvest ~2x106 cells (~1-2 wells of adherent mammalian cells from a 6-well plate) per sample using the standard trypsinization procedure.
- Collect the cells in growth medium and transfer to 15 mL conical tubes.
- Pellet the cells at room temperature at 1000 rpm for 3 min.
- Aspirate off the growth medium, but leave enough to cover the pellets. Flick each tube to break up the pellets. Chill the cells on ice.
- Wash the cells 1x with cold FACS buffer: Add 1 mL cold FACS buffer to each sample. Pellet the cells at room temperature at 1000 rpm for 3 min. Aspirate off the buffer, leaving enough to cover the pellets. Flick each tube to break up the pellets. Resuspend the cells in 1 mL cold FACS buffer.
Bring the ice bucket with the cell samples and cold FACS buffer to your bench. The following steps do not need to be carried out under sterile conditions. However, if you do not have any 1xPBS at your bench, please go to the tissue culture room and make an aliquot in a 50 mL conical in the biosafety cabinet, then keep the 50 mL (labeled "non sterile 1xPBS") at your bench. Do not handle dedicated TC reagents outside of the TC room.
Set up for flow cytometry (single tubes)
- Power-on the flow cytometer and allow it to warm-up as needed.
- Resuspend each cell sample (disrupted cell pellet) in 500 μL of cold FACS Buffer. Keep the samples on ice.
- Label one empty FACS tube with strainer cap per sample and place them on ice.
- Using a 1000 μL micropipette, force each 500 μL of cells through a separate, labeled, clean FACS tube strainer cap.
- Fill an additional FACS tube with ~2 mL clean water.
- Take the following (on ice) to the flow cytometer: (1) strained cell samples in labeled FACS tubes, (2) FACS tube with clean water, (3) left-over FACS buffer.
- Following the manufacturer's software instructions, enter the appropriate settings per these basic, general guidelines:
- Total cell counts: 5,000 - 10,000 is recommended for mammalian cells. Note: For abundant cell samples, higher counts will be achieved in less time, and will use up very little volume of sample.
- Cell size scatter-plot: set up a live display window with forward-scatter and side-scatter on the x- and y-axis.
- Fluorescence signal histogram: set up a live display window with the appropriate fluorescence signal on the x-axis (e.g. mCherry/RFP) and cell counts on the y-axis. Set up one per fluorophore (e.g. one for mCherry/RFP and one for GFP if cells express either or both in your experiment)
- Advanced - Dual signal scatter plot: set up a live display window with one fluorescence signal on the x-axis and the other fluorescence signal on the y-axis.
Run flow cytometry (single tubes)
- Following the manufacturer's instructions, load the clean water tube. Run an analysis until no events appear in the gating displays to ensure that the flow stream is clear.
- Load the "blank" control sample. Run an analysis.
Note: If possible, while the sample is running, adjust the voltage/ signal detection sensitivity so that the peak of blank cells falls at 102 on the x-axis on the Fluorescence signal histogram. This will allow you to compare cells that are both positive and negative for fluorescence. - After the blank sample run is complete, use the software to create threshold gates:
- Cell size scatter-plot - live cell gate: in this window, draw a polygon/ free-form gate around the cloud of dots ("events" or cells) that clusters near the diagonal slope. These are often considered live cells. Use your subjective judgement to exclude the outliers (dead cells and particles) that fall near the x- or y-axis, and have extreme values on the diagonal. See Figure 1 (below) for further explanation.
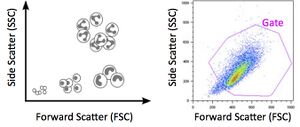
Figure 1. As shown in the cartoon illustration (left), forward scatter correlates with cell size, while side scatter correlates with cell granularity. A FSC/SSC plot (right) can be used to gate a "live" population of cells, i.e. the subgroup with the most typical morphology. - Fluorescence signal histogram - positive fluorescence gate: in this window, create a goal-post or I-beam style gate that excludes the blank cells, and includes the x-axis values where you expect positive values to appear. Note: some experiments allow you to set up a positive control, or include a sample that you know has detectable signal. That sample can be used to edit the positive fluorescence gate later, if you choose.
- Make sure you know how to apply the same gates across all data that will be generated after the blank.
- Cell size scatter-plot - live cell gate: in this window, draw a polygon/ free-form gate around the cloud of dots ("events" or cells) that clusters near the diagonal slope. These are often considered live cells. Use your subjective judgement to exclude the outliers (dead cells and particles) that fall near the x- or y-axis, and have extreme values on the diagonal. See Figure 1 (below) for further explanation.
- Use the software to save the data as "Blank".
- Load the clean water tube. Run an analysis until no events appear in the gating displays to ensure that the flow stream is clear.
- Load the first experimental sample. Run an analysis. Use the software to save the data with an appropriate label.
- If you are running additional samples, clear the flow stream with the clean water sample prior to each cell sample run.

Note: If necessary, dilute the sample with additional cold FACS buffer if the reads per second exceed the maximum recommended limit.
Analyze the data
Instructions and tools vary across different software packages. General guidelines are provided here for your reference
- Limit your fluorescence analysis to the live cell population - if possible, edit the Fluorescence signal histogram - positive fluorescence gate to include only events from the Cell size scatter-plot - live cell gate. This will clean up artifacts (granules and dead cells) from the fluorescence data.
- Layer the histograms - whenever possible display fluorescence histograms (for a single fluor) all on the same plot. This makes the relationship between the independent and dependent variable much clearer.
- Use mean or median fluorescence to compare expression levels - Fluorescence-positive cells peaks (that fall above the blank level) may vary (e.g., 103, 104, 103, etc.). This information shows the differences in the magnitude of fluorescent gene expression. Use only the events in the Fluorescence signal histogram - positive fluorescence gate to calculate a representative signal brightness. Do not include the zero-signal cells (the ones that fall outside the gate).
